


INTRODUCTION
Sepsis is a life-threatening organ dysfunction caused by the imbalance of the host response to infection, and it is one of the leading causes of death in the intensive care unit (ICU).[1,2] Although the treatments of sepsis have been constantly improved in recent years, the mortality rate of sepsis remains high. The liver plays an essential role in the fight against infection and immune defense in sepsis, and liver injury is one of the fatal complications of sepsis.
Autophagy is a highly conserved lysosomal degradation process. Invading microbes, injuried organelles, or abnormal proteins are encapsulated by double membranes to form autophagosomes, which then fuse with lysosomes and are degraded by them. Useful substances are reused for cell metabolism and energy generation.[3.4] Autophagy plays an important role in physiological processes such as cell growth, differentiation, and innate immune defense. It is also involved in inflammatory responses, cancer, and neurodegenerative diseases.[5,6] Recently, autophagy has been reported to be involved in sepsis. However, it is transiently increased and then weakened, accompanied by a specific degree of organ dysfunction.[7] Although the mechanism of autophagy decline in sepsis is still unclear, several studies[8⇓-10] have shown that promoting autophagy can improve organ function and prognosis. Thus, autophagy activation might be a novel therapeutic strategy for sepsis.
Trichostatin A (TSA), a histone deacetylase inhibitor, is a widely used anticancer drug that exerts its antitumor activity by inducing cell cycle arrest, differentiation, and apoptosis.[11] Recently, TSA has been shown to regulate inflammation and autophagy. Kim et al[12] reported that TSA alleviated liver injury by inhibiting the toll-like receptor (TLR)-mediated inflammatory response during sepsis. Cui et al[13] found that TSA could promote M2 polarization of peritoneal macrophages by enhancing autophagy to attenuate inflammation and organ injury (lung, liver and kidney) in sepsis.
Forkhead box O3a (FoxO3a) belongs to the forkhead box O (FoxO) transcription factor family, which plays an important role in the gene regulation of cell growth, proliferation, differentiation, and longevity. Reportedly, FoxO3a regulates inflammation and autophagy. Guo et al[14] found that total flavonoids (TFs) inhibited nuclear factor-kappa B (NF-κB) translocation and activated farnesoid X receptor (FXR) and FoxO3a, which led to a decrease in interleukin-β (IL-β), interleukin-6 (IL-6), high mobility group box-1 (HMGB-1), and cyclooxygenase-2 (COX-2). Liu et al[15] reported that FoxO3a was upregulated by adenosine monophosphate-activated protein kinase (AMPK) to induce autophagy and ameliorate D-galactosamine (D-GalN)/lipopolysaccharide (LPS)-induced acute liver failure.
In summary, autophagy plays a critical role in sepsis. TSA and FoxO3a have the ability to regulate inflammation and autophagy in some diseases. However, the mechanism underlying TSA-mediated improvement in liver injury remains unknown. In this study, we hypothesized that TSA might improve the inflammatory response and liver injury in sepsis through the FoxO3a/autophagy signaling pathway.
METHODS
Establishment of the sepsis mouse model
In this study, a total of 40 male Institute of Cancer Research (ICR) mice of specific-pathogen-free (SPF) grade, aged 8-10 weeks and weighing 25±2 g, were randomly divided into four groups, namely control group, TSA group, CLP group, and CLP+TSA group, with 10 mice in each group. In the control group, the mice only underwent laparotomy but not cecal ligation and puncture (CLP). In the TSA group, the mice were intraperitoneally injected with TSA (2 mg/kg). In the CLP group, the mice underwent CLP. In the CLP+TSA group, the mice were intraperitoneally injected with TSA (2 mg/kg) 30 min before CLP.
The experiment started after one week of adaptive feeding under a 12-h photoperiod. Before the experiment, the mice were fed a regular diet. When the experiment began, they were fasted overnight but allowed to drink water freely. The mice were anesthetized with 2% sodium pentobarbital injected intraperitoneally, the abdomen was shaved and disinfected with alcohol, and a 1-cm longitudinal incision was made in the midline of the abdomen. The cecum was ligated with 5-0 silk thread (from the end of the cecum to the midpoint of the ileocaecal valve). The puncture was performed once with a 21-G needle (to avoid injuring blood vessels, the puncture site was the midpoint of ligation and the end of cecum). After gently squeezing out a small amount of feces, the cecum was placed in the abdominal cavity, and the abdominal incision was sutured. The normal body temperature of the mice was maintained using a heating blanket for small animals during the operation, and 0.5 mL of preheated saline was injected intraperitoneally to supplement the fluid loss during the operation. When the mice woke up on the heating blanket, they returned to the cage and were allowed to drink water freely.
Establishment of the sepsis cell model
AML12 cells were purchased from Saibaikang Biotechnology Co., Ltd. (China) and cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum, 1% penicillin, and 1% streptomycin at 37 °C and 5% CO2 in an incubator under a humidified atmosphere. The cells at the logarithmic growth stage (the 3rd-10th generation) were harvested and placed on the cell culture plate. There were four groups in this cell experiment, namely control group, TSA group, LPS group, and LPS+TSA group. The LPS (1 μg/mL) and TSA (1 μmol/L) concentrations were determined by cell viability experiments. When the cells were 50%-70% confluent, LPS (1 μg/mL) was administered for 12 h to establish the sepsis cell model.
Experimental protocol
The sampling time point of all experiments was 12 h after the intervention (except for specific time regulations). The liver tissue and serum were collected for animal experiments, and the supernatant and cells were collected for cell experiments. The 3-methyladenine (3-MA) (15 mg/kg) was injected intraperitoneally immediately after CLP, and TSA (2 mg/kg) was injected intraperitoneally 30 min before CLP.
Hematoxylin-eosin (H&E) staining
The liver tissues were first immersed in 4% paraformaldehyde for 72 h, and the fixed tissue blocks were sliced into paraffin sections for preservation. For staining, the paraffin sections were dewaxed, stained with H&E, dehydrated, sealed, and observed.
Enzyme-linked immunosorbent assay (ELISA)
The ELISA kit (Senxiong Co., Ltd., China) was maintained at 25 °C for the experiment. All procedures were carried out in accordance with the manufacturer's instructions. The absorbance value of the samples at 450 nm was detected with a microplate reader, and the serum concentrations of IL-6, tumor necrosis factor-α (TNF-α), aspartate transaminase (AST), and alanine aminotransferase (ALT) were calculated according to the protein standard curve.
Transmission electron microscopy
The liver tissue was sliced into sections (3 mm×3 mm×3 mm) and fixed for more than 2 h at 4 °C using glutaraldehyde (2.5%). Then, the sections were immersed in phosphate buffer (0.1 mol/L) overnight, followed by secondary fixation with osmium acid (1%) for 1 h. Subsequently, the sections were washed three times with water for 10 min each, followed by acetone gradient dehydration. Then, the sections were semi-permeabilized with the embedding agent and acetone for 2 h and pure resin permeation for 3 h. The polymerization was carried out for 12 h at 37 °C, 45 °C, and 60 °C each. Finally, the sections were double-stained with lead and uranium and observed by electron microscopy (the protocol was provided by the electron microscopy room of the China-Japan Friendship Hospital).
Immunofluorescence staining
The liver tissues and cells were fixed with 4% paraformaldehyde for 20 min, filtered with 0.2% Triton X-100 for 20 min, and blocked with bovine serum albumin for 60 min. The slides were incubated with sufficient volumes of diluted primary antibody LC3 (1:50, Sigma, USA) and FoxO3a (1:50, Abcam, USA) in a wet box at room temperature for 2 h and then overnight, followed by incubation with the fluorescence secondary antibody (ZSGB-BIO, China) at room temperature for 1 h in the dark for observation.
Western blotting
High-efficiency tissue lysate radioimmunoprecipitation assay (RIPA) buffer was added to the liver tissue on ice for grinding with an electric homogenizer. The supernatant of the tissue lysate was collected by centrifugation. Then, trypsin was added to the pellet to obtain a single-cell suspension. The cell precipitate was washed three times with phosphate-buffered saline (PBS) and resuspended in high-efficiency tissue lysate RIPA buffer. The protein concentration was measured using a bicinchoninic acid (BCA) protein concentration determination kit (Thermo, USA). An equivalent amount of protein was separated by electrophoresis and transferred to 0.2-µm polyvinylidene fluoride (PVDF) membranes (Millipore, USA). The blots were blocked with 5% skimmed milk in Tris-buffered saline with 0.1% Tween 20 (TBST) for 2 h. Then, the blots were probed with primary antibodies against LC3 (Sigma-Aldrich, USA), P62 (Abcam, USA), FoxO3a (Abcam, USA), and β-actin (ZSGB-BIO, China) at 4 °C overnight. Then, the blots were washed three times with TBST for 10 min each and incubated with horseradish peroxidase-labeled secondary antibody (1:500 dilution) at 25 °C for 1 h. Finally, the membrane was washed three times for 10 min each, and the chromogenic agent was added. The immunoreactive bands were visualized on the Bio-Rad gel imager system (Image Lab [version 5.2] software), and the gray value was analyzed. The protein level was normalized to that of β-actin.
Small interfering RNA (siRNA)
The siRNA was synthesized by Gemma gene (China). Cells were transfected with the corresponding siRNA for the FoxO3a gene according to the manufacturer's instructions. Gene silencing efficiency was assessed by Western blotting after 36 h of transfection, and the most efficient construct was used for subsequent experiments.
Statistical analysis
SPSS 21.0 software was used for statistical analysis. Data were presented as the means±standard deviations (SD). For data with normal distribution and homogeneous variances, an independent sample t-test was used for comparison between two groups, and analysis of variance (ANOVA) was used for comparison among groups. A nonparametric statistical rank-sum test was used for data that did not conform to a normal distribution or had uneven variance. The differences were considered statistically significant at a P-value <0.05.
RESULTS
TSA improved liver injury and inflammation in septic mice
To investigate effects of TSA on liver function and inflammation in septic mice, the mice were intraperitoneally injected with TSA (2 mg/kg) 30 min before CLP. Compared with the control group, liver Kupffer cells in the hepatic sinusoids were stimulated and increased, and granulocyte infiltration (Figure 1A), ALT, AST, IL-6, and TNF-α were significantly increased in the CLP group (Table 1), indicating cell injury and inflammation in the livers of septic mice. After TSA intervention, the number of inflammatory cells, ALT, AST, IL-6, and TNF-α decreased significantly in the liver tissue (Table 1), indicating that TSA improved liver injury and alleviated the inflammatory response in septic mice.
Figure 1.
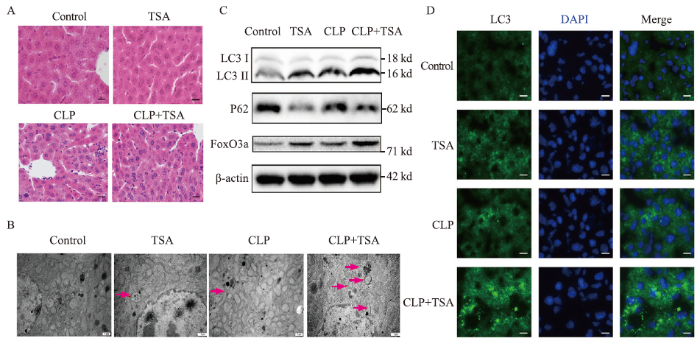
Figure 1.
TSA improved liver injury and inflammation and promoted autophagy and the expression of FoxO3a in the septic mice. A: the liver injury evaluated by H&E staining (200×); B: electron microscope results of the liver tissue in each group after different treatments (12,000×); C: the expression of LC3, P62, and FoxO3a in the liver tissue measured by Western blotting; D: immunofluorescence results of LC3 in the liver tissue after different treatments (400×). TSA: trichostatin A; CLP: cecal ligation and puncture; H&E: hematoxylin-eosin; DAPI: 4',6-diamidino-2-phenylindole.
Table 1. Comparison of ALT, AST, IL-6, TNF-α in the serum and LC3 II, P62, and FoxO3a in the liver tissue at 12 h after the intervention in Institute of Cancer Research (ICR) mice
| Parameters | Control group | TSA group | CLP group | CLP+TSA group |
|---|---|---|---|---|
| ALT a, U/L | 54.92±9.68 | 34.36±12.90 | 198.18±27.07** | 128.42±20.55# |
| AST a, U/L | 206.05±17.11 | 100.49±6.39** | 634.98±74.10** | 478.60±32.56# |
| IL-6 a, pg/mL | 371.62±27.69 | 306.04±13.05** | 2,665.27±324.90** | 2,080.26±373.66# |
| TNF-α a, pg/mL | 177.79±24.72 | 135.00±17.40* | 399.01±60.98** | 221.90±46.89## |
| LC3 II b | 1 | 1.46±0.02** | 1.36±0.01** | 1.66±0.04## |
| P62 b | 1 | 0.60±0.02** | 0.87±0.02** | 0.59±0.02## |
| FoxO3a b | 1 | 2.03±0.13** | 1.49±0.02** | 2.89±0.03## |
The data were represented as mean±standard deviation; a: measured by ELISA in the serum; b: measured by Western blotting; ALT: alanine aminotransferase; AST: aspartate transaminase; IL-6: interleukin-6; TNF-α: tumor necrosis factor-α; FoxO3a: forkhead box O3a; ELISA: enzyme-linked immunosorbent assay. Compared with the control group, *P<0.05, **P<0.01; compared with the CLP group, #P<0.05, ##P<0.01.
TSA promoted liver autophagy and FoxO3a expression in the livers of septic mice
The autophagosome is defined as a double-membrane structure that encloses different stages of degradation of the cytoplasm and injuried organelles. Electron microscopy revealed autophagosomes in both the TSA and CLP groups (red arrow), which were increased in the TSA+CLP group (Figure 1B). Compared with the control group, LC3 II expression was increased and P62 expression was decreased in both the TSA and CLP groups (Figure 1C and Table 1), and LC3 protein fluorescence intensity was increased (Figure 1D), suggesting that TSA promoted autophagy and autophagosomes accumulated in the liver of septic mice. Compared with the CLP group, LC3 II continued to increase, P62 decreased, and LC3 fluorescence intensity continued to increase in the TSA+CLP group, suggesting that TSA promoted liver autophagy in septic mice.
TSA improved inflammation and injury in LPS-stimulated AML12 cells
To validate that TSA regulates liver injury, we used LPS (1 μg/mL)-stimulated AML12 cells. As shown in Figure 2, the IL-6 and TNF-α expression increased at 6-24 h post-LPS stimulation, and TSA suppressed LPS-induced IL-6 and TNF-α expression (Figures 2 A and B). Moreover, the high levels of ALT and AST elevated by LPS stimulation were reduced after TSA treatment (Table 2).
Figure 2.
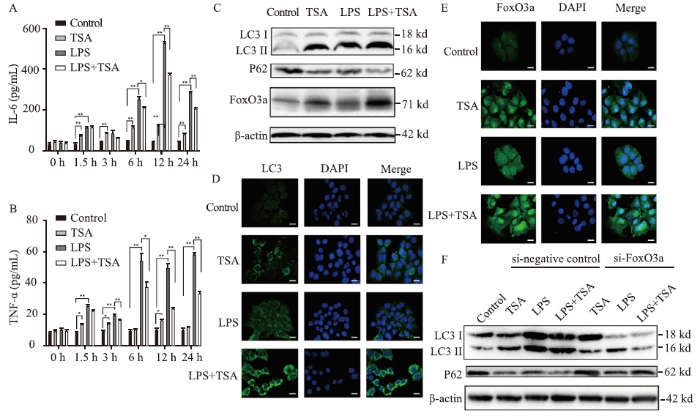
Figure 2.
FoxO3a was involved in the induction of autophagy and the improvement of liver injury and inflammation in the sepsis cell model by TSA. The expression of IL-6 (A) and TNF-α (B) measured by ELISA; the expression of LC3, P62, and FoxO3a analyzed by Western blotting (C); immunofluorescence results of LC3 (D) and FoxO3a (E) detected by confocal microscopy (400×); the changes of LC3 and P62 expression after FoxO3a gene knocked down in AML12 cells in each group analyzed by Western blotting (F). FoxO3a: forkhead box O3a; IL-6: interleukin-6; TNF-α: tumor necrosis factor-α; TSA: trichostatin A; LPS: lipopolysaccharide; ELISA: enzyme-linked immunosorbent assay; DAPI: 4',6-diamidino-2-phenylindole. Compared with control group, *P<0.05, **P<0.01.
Table 2. Comparison of ALT, AST, LC3 II, P62, and FoxO3a in AML12 cells
| Parameters | Control group | TSA group | LPS group | LPS+TSA group |
|---|---|---|---|---|
| ALT a, U/L | 5.05±0.55 | 14.20±2.20 | 29.42±0.62** | 23.47±0.64## |
| AST a, U/L | 9.55±0.65 | 18.90±3.10 | 63.52±4.50** | 38.51±1.50## |
| LC3 II b | 1 | 1.77±0.02** | 1.39±0.01** | 1.87±0.01## |
| P62 b | 1 | 0.50±0.01** | 0.73±0.01** | 0.38±0.01## |
| Foxo3a b | 1 | 2.23±0.02** | 1.79±0.01** | 3.10±0.07## |
The supernatant and cells were collected 12 h after the intervention. The data were represented as mean±standard deviation; a: measured by ELISA; b: measured by Western blotting. ALT: alanine aminotransferase; AST: aspartate transaminase; FoxO3a: forkhead box O3a; ELISA: enzyme-linked immunosorbent assay. Compared with the control group, *P<0.05, **P<0.01; compared with the LPS group, #P<0.05, ##P<0.01.
TSA promoted liver autophagy and FoxO3a expression in LPS-stimulated AML12 cells
Compared with the control group, LC3 II and FoxO3a increased and P62 decreased in the TSA (1 μmol/L) and LPS (1 μg/mL) groups (Figures 2C), and LC3 and FoxO3a fluorescence intensity increased (Figures 2 D and E). Moreover, LC3 II and FoxO3a showed an obvious increase and P62 showed an obvious decrease in the LPS+TSA group (Figure 2C), and LC3 and FoxO3a fluorescence intensity increased markedly (Figures 2 D and E). These data indicated that TSA promoted liver autophagy and FoxO3a expression in LPS-stimulated AML12 cells.
FoxO3a was involved in the induction of autophagy and the improvement of liver injury and inflammation in the sepsis cell model by TSA
In this study, we found that TSA promoted autophagy and FoxO3a expression in LPS-stimulated AML12 cells. To investigate the correlation between TSA and FoxO3a, autophagy, we used siRNA knockdown of FoxO3a in AML12 cells and assessed the correlation between TSA and autophagy, liver function, and inflammation. After inhibition of FoxO3a by si-FoxO3a, low levels of LC3 II and high levels of P62 (Figure 2F), IL-6, and TNF-α were detected in both the LPS and LPS+TSA groups (Table 3). In addition, after knocking down FoxO3a gene, the levels of ALT and AST in both the LPS and LPS+TSA groups showed a significant increasing trend (Table 3; P<0.01).
Table 3. Comparison of ALT, AST, IL-6, TNF-α, LC3 II, and P62 after FoxO3a gene knocked down in AML12 cells among groups
| Parameters | Control group | si-negative control | si-FoxO3a | ||||
|---|---|---|---|---|---|---|---|
| TSA group | LPS group | LPS+TSA group | TSA group | LPS group | LPS+TSA group | ||
| ALT a, U/L | 5.37±0.63 | 14.13±1.80 | 29.27±0.52 | 23.33±0.50 | 14.63±1.03 | 38.57±2.30** | 29.20±1.10** |
| AST a, U/L | 9.07±0.87 | 20.27±3.18 | 57.00±1.59 | 39.57±1.94 | 19.63±3.03 | 71.23±5.85* | 46.70±2.90* |
| IL-6 a, pg/mL | 40.05±1.96 | 111.77±2.53 | 500.50±7.96 | 355.07±9.71 | 132.07±2.76** | 598.57±14.01** | 413.73±26.24* |
| TNF-α a, pg/mL | 10.33±1.25 | 11.93±1.69 | 54.15±2.79 | 36.77±2.39 | 13.47±1.92* | 66.47±3.03* | 50.43±3.35** |
| LC3 II b | 1 | 1.51±0.01 | 2.51±0.02 | 2.57±0.01 | 1.22±0.01** | 1.69±0.01** | 1.28±0.04** |
| P62 b | 1 | 0.48±0.01 | 0.54±0.01 | 0.33±0.01 | 0.94±0.01** | 0.66±0.01** | 0.93±0.02** |
The supernatant and cells were collected 12 h after the intervention. The data were represented as mean±standard deviation; a: measured by ELISA; b: measured by Western blotting. ALT: alanine aminotransferase; AST: aspartate transaminase; IL-6: interleukin-6; TNF-α: tumor necrosis factor-α; FoxO3a: forkhead box O3a; ELISA: enzyme-linked immunosorbent assay; si-FoxO3a: siRNA knockdown of FoxO3a. Compared with the si-negative control group, *P<0.05, **P<0.01.
DISCUSSION
In 2016, the definition of sepsis was updated as life-threatening organ dysfunction. The immune response caused by invasive pathogens does not restore homeostasis, leading to persistent excessive inflammation, immunosuppression, and ultimately organ dysfunction.[16]Although the “Save Sepsis” campaign guide is being used worldwide to reduce sepsis deaths, it is not sufficient. Thus, exploring the pathophysiological mechanism of sepsis is essential. The CLP model is the most widely used experimental sepsis model.[17]The mortality in the model is associated with the length of cecal ligation, the size of the needle, and the number of punctures. In the present study, 50% cecal ligation and 21-G needle puncture were used to construct a model of intermediate sepsis. The model was considered successful when mice showed symptoms, such as fever, chills, reduced activity, elevated inflammatory factors, and severe liver injury. LPS is a major component of the cytoderm of Gram-negative bacteria and the leading cause of sepsis. Here, sepsis cell models were established by LPS intervention in AML12 cells.
During sepsis, LPS interacts with toll-like receptor 4 (TLR4) to activate the MyD88/NF-κB pathway and induce the synthesis and release of TNF-α, IL-1β, IL-6, and other proinflammatory cytokines,[18] which in turn promotes the TLR4 pathway that drives the inflammatory response into a vicious cycle.[19] Elevated levels of TNF-α and IL-6 are biomarkers of poor prognosis of sepsis.[20] The liver is involved in the regulation of sepsis inflammation due to its abundance of macrophages and extensive endothelial surface, but it is also a victim of inflammation. Thus, hepatic failure is strongly associated with high mortality in sepsis.[12]
In this study, ALT and AST levels were significantly increased, and inflammatory cell infiltration in the liver tissue, IL-6, and TNF-α was significantly increased in the CLP group. A similar trend in ALT, AST, TNF-α, and IL-6 levels was observed in LPS-stimulated AML12 cells. However, ALT, AST, TNF-α, and IL-6 levels decreased significantly after TSA intervention in both septic mice and AML12 cells.
TSA regulates macrophage phenotypes by enhancing autophagy, which reduces inflammation in multimicrobial sepsis.[13]According to the current evidence, autophagy is an essential component of the innate immune response. In sepsis, autophagy protects organ function by clearing invading microbes, removing injuried organelles, and regulating the production and release of cytokines, thereby reducing proinflammatory signals.[21⇓-23] Autophagy involves a synergy of various autophagy-related proteins, of which LC3 and P62 localization is crucial. The transformation of LC3 from LC3 I (free form) to LC3 II (membrane-bound form) is considered an essential step in autophagy, and LC3 II is used as a marker molecule to reflect the number of autophagosomes. The P62 protein connects the ubiquitinated protein to LC3 in the autophagosome membrane and is degraded in the process. In the current study, after TSA intervention, LC3 II expression was increased significantly, and P62 expression was decreased, suggesting that TSA promoted the formation of autophagosomes that accumulated in the livers of septic mice. Moreover, we found that when septic mice were treated with the autophagy blocker 3-MA, the LC3 II protein level was decreased, while P62 accumulated, and liver function and inflammation were deteriorated (supplementary Figure 1). Therefore, autophagy plays an essential role in liver injury in sepsis. In the septic animal models, after TSA intervention, the number of autophagosomes, LC3 II expression, and FoxO3a levels were increased, while P62 expression was decreased. These findings suggested that TSA promoted the formation of autophagosomes in hepatocytes of septic mice.
FoxO3a, a member of the FoxO family, is a transcription factor that regulates the growth, survival, and aging of mammalian cells. Accumulating evidence indicates that FoxO3a is involved in autophagy. Melatonin prevents neuroinflammation and alleviates depression by regulating FoxO3a and reducing autophagy disorders.[24]Additionally, TSA regulation of FoxO3a has been reported. TSA alleviates oxidative stress-mediated myocardial injury in rats through the FoxO3a signaling pathway,[25] activates protein kinase B (AKT)/FoxO3a, reduces the generation of the proapoptotic protein BIM, alleviates myocardial ischemia-reperfusion injury in diabetic rats,[26] and promotes FoxO3a expression. Herein, we observed that TSA promoted FoxO3a expression in septic mice and AML12 cell models. We used siRNA to knock down the FoxO3a gene in cells. Compared with the nonknockdown group, TSA reduced the promotion of autophagy and weakened the improvement in the liver enzyme index and inflammation. These findings suggested that FoxO3a participated in the regulation of autophagy by TSA and the inflammatory response and liver injury in sepsis. Notably, FoxO3a fluorescence was concentrated in the nucleus after TSA intervention. Based on previous studies,[11,26] we speculated that TSA regulated FoxO3a promoter acetylation and/or FoxO3a acetylation, resulting in increased FoxO3a expression and aggregation in the nucleus and strong transcriptional activity.
CONCLUSIONS
The current study provides new insights into TSA-regulated septic liver injury and inflammation. The inhibition of autophagy aggravates septic liver injury, and TSA promotes autophagy to improve liver injury and inflammation in septic mice. Although this phenomenon was observed only in animal and cell models, the results may be valuable for the development of TSA as a novel therapeutic drug for inflammatory diseases. Based on the current results, it is necessary to explore the regulatory mechanism of TSA in autophagy in sepsis for future application.
Funding: This study was supported by a grant from National Natural Science Foundation of China (81871600).
Ethical approval: All operations performed on the animals in this experiment were approved by the Ethics Committee of China-Japan Friendship Clinical Medicine Institute of China National Health Commission (Ethics number: 180102) according to the relevant guidelines.
Conflicts of interests: There are no financial or other conflicts of interests related to this article.
Contributors: MJS proposed and wrote the paper. All authors read and approved the final version.
The supplementary file in this paper is available at http://wjem.com.cn.
Reference
Improving mortality in sepsis: analysis of clinical trials
DOI:10.1097/SHK.0b013e3181e7e8b4 URL [Cited within: 1]
Comparison of mortality associated with sepsis in the burn, trauma, and general intensive care unit patient: a systematic review of the literature
DOI:10.1097/SHK.0b013e318237d6bf URL [Cited within: 1]
Autophagosome formation in mammalian cells
DOI:10.1247/csf.27.421
PMID:12576635

Macroautophagy is an intracellular degradation system for the majority of proteins and some organelles. The molecular mechanism of autophagy has been extensively studied using the yeast, Saccharomyces cerevisiae, during these past 10 years. These studies suggested that the molecular machinery of autophagosome formation is well conserved from yeast to higher eukaryotes. Identification and characterization of the mammalian counterparts of the yeast autophagy proteins has facilitated our understanding of mammalian autophagy, particularly of autophagosome formation. These findings are now being applied to studies on the physiological roles of autophagy in mammals.
Autophagy in immunity and inflammation
Autophagy in the pathogenesis of disease
DOI:10.1016/j.cell.2007.12.018
PMID:18191218
[Cited within: 1]
Autophagy is a lysosomal degradation pathway that is essential for survival, differentiation, development, and homeostasis. Autophagy principally serves an adaptive role to protect organisms against diverse pathologies, including infections, cancer, neurodegeneration, aging, and heart disease. However, in certain experimental disease settings, the self-cannibalistic or, paradoxically, even the prosurvival functions of autophagy may be deleterious. This Review summarizes recent advances in understanding the physiological functions of autophagy and its possible roles in the causation and prevention of human diseases.
Autophagy in mammalian development and differentiation
DOI:10.1038/ncb0910-823
PMID:20811354
[Cited within: 1]
It has been known for many decades that autophagy, a conserved lysosomal degradation pathway, is highly active during differentiation and development. However, until the discovery of the autophagy-related (ATG) genes in the 1990s, the functional significance of this activity was unknown. Initially, genetic knockout studies of ATG genes in lower eukaryotes revealed an essential role for the autophagy pathway in differentiation and development. In recent years, the analyses of systemic and tissue-specific knockout models of ATG genes in mice has led to an explosion of knowledge about the functions of autophagy in mammalian development and differentiation. Here we review the main advances in our understanding of these functions.
The decline of autophagy contributes to proximal tubular dysfunction during sepsis
DOI:10.1097/SHK.0b013e318240b52a URL [Cited within: 1]
IL-22 ameliorates LPS-induced acute liver injury by autophagy activation through ATF4-ATG7 signaling
DOI:10.1038/s41419-020-03176-4 URL [Cited within: 1]
Hepatoprotective effect of anemoside B4 against sepsis-induced acute liver injury through modulating the mTOR/p70S6K-mediated autophagy
DOI:10.1016/j.cbi.2021.109534 URL [Cited within: 1]
Dexmedetomidine protects against septic liver injury by enhancing autophagy through activation of the AMPK/SIRT1 signaling pathway
DOI:10.3389/fphar.2021.658677 URL [Cited within: 1]
Histone deacetylase inhibitors induce autophagy through FoxO1-dependent pathways
DOI:10.1080/15548627.2015.1023981 URL [Cited within: 2]
Trichostatin A protects liver against septic injury through inhibiting toll-like receptor signaling
DOI:10.4062/biomolther.2015.176 URL [Cited within: 2]
Trichostatin A modulates the macrophage phenotype by enhancing autophagy to reduce inflammation during polymicrobial sepsis
DOI:10.1016/j.intimp.2019.105973 URL [Cited within: 2]
Trichostatin A attenuates oxidative stress-mediated myocardial injury through the FoxO3a signaling pathway
DOI:10.3892/ijmm.2017.3101 URL [Cited within: 1]
AMPK activation ameliorates D-GalN/LPS-induced acute liver failure by upregulating FoxO3a to induce autophagy
DOI:10.1016/j.yexcr.2017.07.008 URL [Cited within: 1]
Sepsis and septic shock: a history
DOI:10.1016/j.ccc.2008.12.003 URL [Cited within: 1]
Rodent models of intra-abdominal infection
DOI:10.1097/01.shk.0000191386.18818.0a URL [Cited within: 1]
Introduction to NF-kappaB: players, pathways, perspectives
DOI:10.1038/sj.onc.1209954
PMID:17072321
[Cited within: 1]
This article serves as an introduction to the collection of reviews on nuclear factor-kappaB (NF-kappaB). It provides an overview of the discovery and current status of NF-kappaB as a research topic. Described are the structures, activities and regulation of the proteins in the NF-kappaB family of transcription factors. NF-kappaB signaling is primarily regulated by inhibitor kappaB (IkappaB) proteins and the IkappaB kinase complex through two major pathways: the canonical and non-canonical NF-kappaB pathways. The organization and focus of articles included in the following reviews are described, as well as likely future areas of research interest on NF-kappaB.
Pattern recognition receptors and inflammation
DOI:10.1016/j.cell.2010.01.022
PMID:20303872
[Cited within: 1]
Infection of cells by microorganisms activates the inflammatory response. The initial sensing of infection is mediated by innate pattern recognition receptors (PRRs), which include Toll-like receptors, RIG-I-like receptors, NOD-like receptors, and C-type lectin receptors. The intracellular signaling cascades triggered by these PRRs lead to transcriptional expression of inflammatory mediators that coordinate the elimination of pathogens and infected cells. However, aberrant activation of this system leads to immunodeficiency, septic shock, or induction of autoimmunity. In this Review, we discuss the role of PRRs, their signaling pathways, and how they control inflammatory responses.2010 Elsevier Inc. All rights reserved.
Successful treatment of acute lung injury with pitavastatin in septic mice: potential role of glucocorticoid receptor expression in alveolar macrophages
DOI:10.1124/jpet.110.171462 URL [Cited within: 1]
Pattern recognition receptors and autophagy
Mitochondrial dysfunction in Sepsis
DOI:10.1007/s11908-003-0015-9
PMID:13678565
[Cited within: 1]
Sepsis is an increasingly common problem, particularly among critically ill patients. Mechanisms by which sepsis induces organ dysfunction have not been elucidated. The coexisting findings (unique to sepsis) of metabolic acidosis yet increased tissue oxygen tensions suggest cellular availability but decreased use of oxygen (tissue dysoxia). Because mitochondria use more than 90% of total body oxygen consumption for adenosine triphosphate (ATP) generation, a bioenergetic abnormality is implied. Cell and animal data have shown that nitric oxide (and its metabolites), produced in considerable excess in patients with sepsis, can affect oxidative phosphorylation by inhibiting several of its component respiratory enzymes. Human data are scarce. However, in skeletal muscle biopsies taken from patients with sepsis, we have recently demonstrated a relationship between increased nitric oxide production, antioxidant depletion, reduced respiratory chain complex I activity, and low ATP levels. These findings correlated with severity of disease and outcome and support the notion that mitochondrial dysfunction resulting in bioenergetic failure may be an important factor in the pathophysiology of sepsis-associated multiorgan failure. However, a reasonable argument can be made that the reduction in energy supply could represent a last-ditch adaptive response to ongoing inflammation, resulting in a cellular shutdown analogous to hibernation that allows eventual restoration of organ function and long-term survival in patients fit enough to survive the acute phase.
Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction
DOI:10.1038/ni.2215 URL [Cited within: 1]
Trichostatin A attenuates oxidative stress-mediated myocardial injury through the FoxO3a signaling pathway
DOI:10.3892/ijmm.2017.3101 URL [Cited within: 1]
Melatonin prevents neuroinflammation and relieves depression by attenuating autophagy impairment through FoxO3a regulation
Suppression of excessive histone deacetylases activity in diabetic hearts attenuates myocardial ischemia/reperfusion injury via mitochondria apoptosis pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}