


INTRODUCTION
Sudden cardiac arrest (CA) is critical for its high morbidity and mortality worldwide. Neurological injury after CA remains a major cause of poor prognosis among survivors.[1,2] Brain injury occurs not only during CA and resuscitation, but also after the reestablishment of brain reperfusion, namely ischemia/reperfusion (I/R) injury, which is a main target of neuroprotective treatments. Unfortunately, the treatment remains suboptimal. Cerebral I/R injury after the successful return of spontaneous circulation (ROSC) is a process driven by multiple mechanisms, including calcium overload, apoptosis, oxidative stress, and inflammation.[3] These mechanisms intersect with each other, discounting the effect of any single pharmacotherapy, even comprehensive interventions (like hypothermia).[4,5] Therefore, it is urgent to find a new, stable, and effective treatment.
Multipotent mesenchymal stem cells (MSCs) play a role in neurovascular remodeling and neurological recovery following cerebral ischemia injury.[6,7] Rather than directly replacing parenchymal brain cells, the therapeutic mechanism of MSCs is suggested to produce extracellular vesicles (EVs) in a paracrine pattern. Small extracellular vesicles (sEVs), referring to 30-200 nm EVs, mainly including exosomes and microvesicles, are lipid bilayer-enclosed structures which contain a variety of cargos such as proteins, lipids, and DNA and RNA species, and participate in cell-to-cell signaling processes.[8] Previous studies have shown that sEVs from the bone marrow mesenchymal stem cells (BMSC-sEVs) play a protective role in myocardial and renal I/R injury.[9,10] However, how sEVs from MSCs protect the neuron cells in I/R is still not clear. In this study, we used oxygen-glucose deprivation and reperfusion (OGD/R) to establish a model of I/R in rat primary cortical neurons. Based on this model, we examined whether the mechanism through which BMSC-sEVs could rescue OGD/R-induced neuronal injury.
METHODS
Animals
Three-week-old male Sprague-Dawley (SD) rats weighing 50-60 g and fetal rats on embryonic days 17 and 18 (E17-18) were purchased from the Animal Experimental Center, Nanjing Medical University, China. All animal experiments were approved by the Institutional Animal Care and Use Committee (approval number: 1801008) of Nanjing Medical University, and carried out following the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Cell isolation and culture
BMSCs were harvested from the three-week-old SD rats. Briefly, the animals were given an intraperitoneal injection of chloral hydrate (300 mg/kg) for anesthesia before they were sacrificed. The marrow was flushed out after removing the epiphysis of femurs and tibias, followed by resuspension in low glucose Dulbecco’s modified Eagle’s medium (L-DMEM) (Gibco, USA). Twenty-four hours later, the medium was replaced to remove the non-adherent cells. Thereafter, the cells were harvested when they achieved 80%-90% confluence, followed by cell passage at a rate of 1:2. BMSCs from 3-4 passages were used for subsequent experiments.
Primary cortical neurons were cultured according to the previous description.[11] In brief, cerebral cortices were collected from the 17- to 18-day rat embryos. Dissected cortices were dissociated and suspended in the neurobasal medium containing 2 mmol glutamine and B27 (Invitrogen, USA), and then inoculated onto the plates coated with poly-L-lysine. All cells were cultured in an incubator with a humidified atmosphere of 5% CO2 at 37 °C. The experiments were carried out at 8-9 days after the initial plating of cultures.
BMSC-sEVs purification and identification
BMSCs from the 3rd and the 4th passages at 70%-80% confluence were washed with phosphate-buffered saline (PBS) before incubation in L-DMEM containing 10% exosome-depleted fetal bovine serum (System Biosciences, USA). Forty-eight hours later, the medium was collected, while sEVs were isolated from the medium by multi-step centrifugation at 4 °C. In brief, the supernatants were filtered using a 0.22-μm filter, centrifuged for 30 minutes at 10,000g and ultra-centrifuged for 70 minutes at 110,000g (Beckman SW32 Ti) to pellet the sEVs. Finally, the sEVs were subjected to resuspension within 100 μL PBS and preserved under the temperature of -80 °C prior to utilization. The concentration of sEVs was determined through evaluating the contents of total protein by bicinchoninic acid (BCA) assay (Sigma-Aldrich, USA). The nanoparticle tracking analysis (NTA) (Malvern, UK) and the electron microscope were used to detect the BMSC-sEVs concentration, size distribution, and their specific markers CD9. Alix and heat shock protein 70 (HSP70) (Abcam, UK) were examined using the Western blot assay.
Internalization of BMSC-sEVs into cortical neurons
BMSC-sEVs were subjected to 5 minutes of PKH26 (Sigma-Aldrich, USA) labeling under the temperature of 37 °C in the dark in accordance with manufacturer protocols, and washed twice in PBS with centrifugation at 110,000g at 4 °C for 2 hours to remove unbound PKH26. Thereafter, the labeled sEVs (40 μg/mL) were added to the prepared cortical neurons for 24 hours. The nuclei were dyed using Hoechst33342 (Beyotime, China). The uptake of BMSC-sEVs by neurons was observed using a laser scanning confocal microscope (Olympus, Japan).
OGD/R
To achieve OGD, neurons were rinsed twice with PBS, cultured in glucose-free DMEM (Gibco, USA), and then put into an incubator containing 95% N2 and 5% CO2 at 37 °C for 2 hours. Then the cultures were incubated again in a normoxic incubator with normal culture medium for an additional 24 hours at 37 °C as reoxygenation (R). Cells were divided into four groups, including the control group, OGD/R group, and OGD/R+sEVs groups (sEVs 20 μg/mL or 40 μg/mL). sEVs were added when initiating the reoxygenation process. Cells that were not exposed to OGD/R were defined as the control group.
Cell viability and lactate dehydrogenase (LDH) assays
After exposure to OGD/R, the cell viability was tested by cell counting kit-8 (CCK-8) (Beyotime, China), and the optical density was detected at the wavelength of 450 nm. The LDH cytotoxicity assay kit (Beyotime, China) was used to quantitatively evaluate neuron damage. Following OGD/R, the culture medium of cortical neurons was centrifuged to obtain the supernatants. The amount of LDH leakage was measured according to the manufacturer’s instructions. The absorbance of the samples was measured spectrophotometrically at 490 nm. The results were expressed as the relative percentage of the control group.
Determination of mitochondrial membrane potential (MMP)
The tetrachloro-tetraethyl benzimidazol carbocyanine iodide fluorescent dye (JC-1) (Beyotime, China) was used to detect MMP. Briefly, cells were subjected to 20 minutes of JC-1 incubation in the dark at 37 °C. Meanwhile, the laser scanning confocal microscope was adopted to capture images. The ratio of JC-1 aggregates (red fluorescence) to monomers (green fluorescence) was calculated using ImageJ (NIH, USA). The loss of mitochondrial function was indicated by a decrease in the ratio of the red/green fluorescence intensity.[12]
Measurement of reactive oxygen species (ROS) generation, superoxide dismutase (SOD), and glutathione peroxidase (GPx)
Intracellular ROS was detected by an antioxidation-sensitive fluorescent probe 2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA) (Beyotime, China). The neurons were washed and then incubated with 10 μmol DCFH-DA at 37 °C for 20 minutes with gentle shaking. The fluorescence intensity was quantified using a fluorospectrophotometer at an excitation wavelength of 485 nm and an emission wavelength of 525 nm. After exposure to OGD/R, the neurons were harvested, sonicated, and centrifuged to collect the supernatants. The levels of SOD and GPx were determined with the respective assay kits (Beyotime, China) according to manufacturers’ instructions.
Measurement of intracellular calcium concentration ([Ca2+]i)
Neurons were washed by PBS and incubated with the complete medium containing 5 μmol Fluo-4 acetoxymethyl (AM) (KeyGen Biotech, China) for 30 minutes in the dark at 37 °C. Subsequently, the cells were washed with PBS and incubated at 37 °C for another 10 minutes prior to measurement. Finally, the fluorescence was analyzed by flow cytometry (BD Biosciences, USA) at an excitation wavelength of 488 nm and an emission wavelength of 530 nm, and the relative mean fluorescence intensity of Fluo-4 was used to indicate the [Ca2+]i.
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) staining
After treatment, neurons were stained with TUNEL dye (Roche, USA) according to the manufacturer’s recommendations. Then, 4',6-diamidino-2-phenylindole (DAPI) (Beyotime, China) was used as the nuclear counterstain. Fluorescence images were acquired with the laser scanning confocal microscope (Olympus, Japan), and TUNEL-positive nuclei of five non-overlapping fields per coverslip were counted. The apoptotic index was expressed as the percentage of the ratio of TUNEL-positive nucleus count to the total nucleus number determined by DAPI counterstaining.
Western blot analysis
Equal amounts of protein were loaded onto 12% sodium dodecyl sulfate-polyacrylamide gels and transferred onto the polyvinylidene fluoride (PVDF) membranes (Millipore Corporation, USA). Afterwards, the membranes were blocked and followed by incubation with primary antibodies at 4 °C overnight. The primary antibodies used were anti-cleaved caspase-3 antibody (1:1,000; Cell Signaling Technology, USA), anti-B-cell lymphoma 2 (Bcl-2) (Abcam, UK), anti-Bcl-2-associated X (Bax) (Cell Signaling Technology, USA), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Cell Signaling Technology, USA), anti-calcium/calmodulin-dependent kinase II (CaMK II) (Cell Signaling Technology, USA), and anti-phosphorylated CaMK II (p-CaMK II) (Abcam, UK). Then, the membranes were further incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies at room temperature for 2 hours, and the resultant protein bands were visualized by enhanced chemiluminescence (Beyotime, China).
Statistical analysis
Data were expressed as the mean±standard deviation (SD) for all parameters. Differences between the two groups were analyzed by Student’s t-test. Multiple comparisons were performed with one-way analysis of variance (ANOVA) followed by the Tukey post-hoc test. All calculations were performed using GraphPad Prism software version 6.0 (GraphPad, CA, USA). The P-value <0.05 was considered statistically significant.
RESULTS
Identification of BMSCs and BMSC-sEVs
The BMSCs appeared as fibroblast-like and spindle-shaped swirling adherent cells under the phase-contrast microscope (Figure 1A). For assessing their differentiation, the cells were induced by the specific medium. BMSCs successfully differentiated into osteoblasts and adipocytes at three to four weeks after induction by osteogenic medium and adipogenic medium, respectively (Figure 1B). The results of transmission electron microscopy showed that the isolated sEVs displayed a bilayer membrane (Figure 1C). The NTA showed that the diameters of the particles were within the range of 50-150 nm, averaging 104 nm (Figure 1D). Furthermore, three specific surface markers (Alix, Hsp70, and CD9) were detected in BMSC-sEVs (Figure 1E). The PKH26 (red)-labeled sEVs swarmed into the neurons to the perinuclear cytoplasm within 24 hours (Figure 1F).
Figure 1.
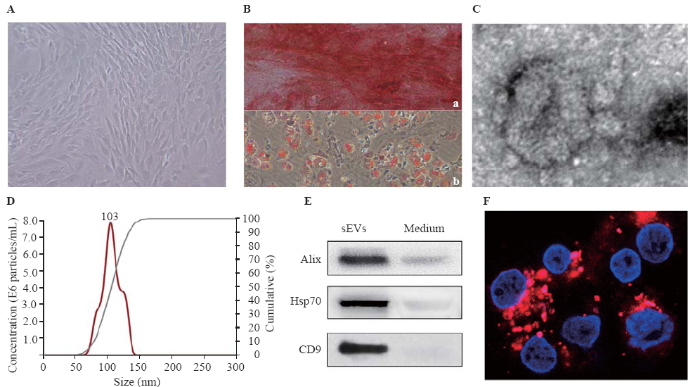
Figure 1.
Identification of BMSCs and BMSC-sEVs. A: morphology of BMSCs under phase-contrast microscope; B: multi-differentiation potential of BMSCs; a: Alizarin Red staining of osteogenic mineralization; b: Oil Red O staining of small lipid droplets (bar=100 μm); C: transmission electron micrograph of BMSC-sEVs (bar=50 nm); D: nanoparticle tracking analysis of sEVs; E: the expression of Alix, Hsp70, CD9 in conditioned medium and sEVs by Western blot assay; F: the uptake of PKH26-labeled sEVs (red) by neurons (4',6-diamidino-2-phenylindole [DAPI] blue) at 24 hours (bar=20 μm); BMSCs: bone marrow mesenchymal stem cells; sEVs: small extracellular vesicles.
BMSC-sEVs protected primary cortical neurons against OGD/R-induced injury
The OGD/R exposure significantly decreased the cell viability and increased the LDH leakage, which was strikingly attenuated by BMSC-sEVs, suggesting that BMSC-sEVs can dependently attenuate OGD/R-induced cell injury in primary cortical neurons (Figures 2A, B).
Figure 2.
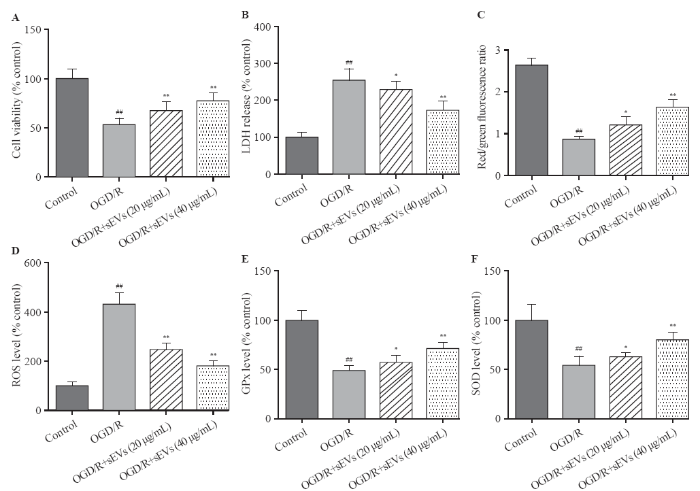
Figure 2.
Effects of BMSC-sEVs on OGD/R-induced neuronal injury and oxidative stress. A: cell viability measured by cell counting kit-8 assay; B: cytotoxicity assessed by lactate dehydrogenase (LDH) assay; C, D, E, and F: levels of oxidative status markers including mitochondrial membrane potential (MMP), reactive oxygen species (ROS) generation, glutathione peroxidase (GPx), and superoxide dismutase (SOD) determined using respective assay kits; BMSCs: bone marrow mesenchymal stem cells; sEVs: small extracellular vesicles; OGD/R: oxygen-glucose deprivation and reperfusion; compared with the control group, ##P<0.01; compared with the OGD/R group, *P<0.05, **P<0.01.
BMSC-sEVs alleviated OGD/R-induced oxidative stress in primary cortical neurons
The OGD/R exposure dramatically reduced the red/green fluorescence intensity of JC-1, suggesting a significant decrease of MMP, but this decrease was inhibited after the treatment of BMSC-sEVs (Figure 2C). Additionally, OGD/R exposure resulted in a 4-fold increase in cellular ROS level. Compared to the control group, the levels of SOD and GPx decreased in OGD/R group, and as speculated, BMSC-sEV treatment obviously attenuated OGD/R-induced ROS generation, and enhanced SOD and GPx activities in rat primary cortical neurons. Taken together, BMSC-sEV treatment can suppress OGD/R-induced oxidative stress in vitro (Figures 2D, E, and F).
BMSC-sEVs reduced OGD/R-induced apoptosis in primary cortical neurons
The TUNEL staining assay was performed to assess the percentage of apoptotic cells. After OGD/R exposure, the TUNEL-positive cells in OGD/R group were significantly increased, while BMSC-sEVs substantially attenuated OGD/R-induced apoptosis, as evidenced by the diminishing TUNEL-positive cells (Figures 3A, B). The protein levels of the apoptosis-associated cleaved caspase-3, Bcl-2, and Bax were measured by Western blot assay. OGD/R increased the cleaved caspase-3 level and decreased the Bcl-2/Bax ratio. These changes were remarkably reversed by BMSC-sEVs, manifested as rising Bcl-2/Bax ratio and falling cleaved caspase-3/GAPDH ratio (Figure 3C).
Figure 3.
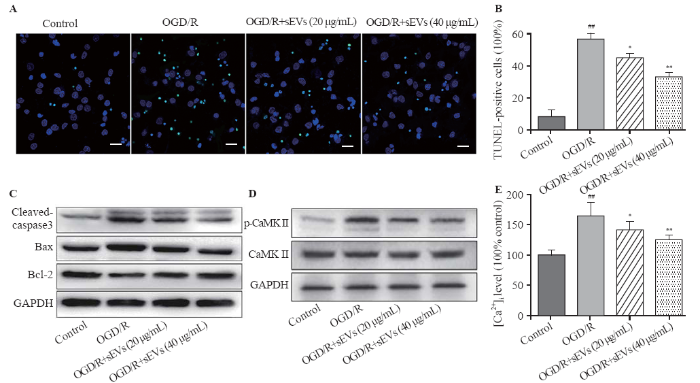
Figure 3.
Effects of BMSC-sEVs on OGD/R-induced neuronal apoptosis and Ca2+/CaMK II signaling pathway. Representative TUNEL images (A) and quantification of TUNEL-positive cells (B) (bar=20 μm); C: the protein levels of cleaved caspase-3, Bax and Bcl-2 determined by Western blot assay; D: the protein levels of CaMK II and p-CaMK II were determined by Western blot assay; E: the relative fluorescent intensity of Fluo-4 used to indicate the [Ca2+]i quantity; BMSCs: bone marrow mesenchymal stem cells; sEVs: small extracellular vesicles; OGD/R: oxygen-glucose deprivation and reperfusion; compared with control group, ##P<0.01; compared with OGD/R group, * P<0.05, **P<0.01.
BMSC-sEV treatment suppressed OGD/R-induced Ca2+/CaMK II signaling in primary cortical neurons
We explored whether Ca2+/CaMK II signaling pathway was involved in the neuroprotective effects of BMSC-sEVs. Western blot analysis showed that the level of p-CaMK II protein was up-regulated by OGD/R exposure, but was dose-dependently down-regulated by BMSC-sEV treatment at concentrations of 20 and 40 μg/mL (Figure 3D). At the same time, BMSC-sEVs suppressed OGD/R-induced elevation of [Ca2+]i (Figure 3E), implying that sEVs may reduce OGD/R-induced Ca2+/CaMK II activation in cortical neurons.
DISCUSSION
A previous study[13] has confirmed that instead of directly reaching the locus of brain injury, MSCs mainly play their therapeutic role relying on their paracrine properties. SEVs have been recognized as important messengers in intercellular communication that act on target cells via transporting bioactive lipids, proteins, and RNAs.[8,14] BMSC-sEVs have been identified as neuroprotective candidates in hypoxia-ischemia brain disease by several studies,[15,16] but no mechanisms have been clarified to explain the protective effects of BMSC-sEVs on I/R-induced neuronal injury.
Brain damages, caused by CA and ROSC, including complete temporary global cerebral ischemia and secondary I/R injury, are complex processes associated with oxidative stress, intracellular Ca2+ overload, inflammation, and apoptosis, all leading to cell death. In our study, BMSC-sEVs attenuated OGD/R-induced neuronal viability and inhibited LDH release, indicating that BMSC-sEVs have a protective effect on OGD/R-induced neuronal damage. Our data also showed that BMSC-sEV treatment significantly antagonized cell injury under OGD/R by inhibiting cell apoptosis. Mitochondria are the main organella initiating oxidative stress. Mitochondrial dysfunction is manifested as the decrease in MMP and the overproduction of ROS.[17] Our data demonstrated that the treatment with BMSC-sEVs suppressed oxidative stress by decreasing ROS production, increasing SOD activity, and strengthening GPx activity in primary rat cortical neurons after OGD/R. These findings are similar to a previous study in that BMSC-sEVs can protect H9C2 cardiomyocytes against H2O2-induced I/R injury by attenuating ROS production and apoptosis.[9] Furthermore, mitochondrial depolarization was observed in neurons after OGD/R, and BMSC-sEVs prevented OGD/R-induced MMP dissipation. Thus, we may conclude that the protective effects of BMSC-sEVs against OGD/R-induced neuron injury are dependent on enhanced mitochondrial function, anti-oxidative stress, and anti-apoptosis.
To further explore the signaling pathways in the neuroprotective effects of BMSC-sEVs, we examined the level of [Ca2+]i and activation of CaMK II protein. CaMK II, a major highly expressed multifunctional Ser/Thr kinase in neuronal tissues, plays a crucial role in a variety of processes, including oxidative stress,[18] apoptosis,[19] axonal and dendritic arborization as well as synaptogenesis.[20] As a second messenger, Ca2+ is involved in numerous cellular processes; therefore, Ca2+ signaling disturbance may evoke neuronal damage.[21] When the [Ca2+]i increases, Ca2+ binds to calmodulin (CaM), and then Ca2+/CaM complex interacts with target proteins to initiate various processes, like the activation of CaMK II.[22] As shown in our study, OGD/R exposure significantly increased the levels of p-CaMK II/CaMK II, but the administration of BMSC-sEVs suppressed the levels of p-CaMK II/CaMK II. This finding was consistent with that of a previous study, which showed that decreasing the expression of p-CaMK II could counter neuronal injury induced by I/R in vivo.[23] Considering the association between CaMK II and [Ca2+]i, we used the Fluo-4 AM probe to detect Ca2+ fluorescence intensity in neurons, and found that Ca2+ fluorescence intensity increased in response to oxidative stress under OGD/R, and decreased after BMSC-sEV treatment. This finding supported the anti-apoptotic effects of BMSC-sEVs on cardiac stem cells in that BMSC-sEVs could restrain oxidative damage and apoptosis by targeting Ca2+/CaMK II.[24] Our data revealed that BMSC-sEV treatment suppressed OGD/R-induced oxidative stress and apoptosis via Ca2+/CaMK II pathway in primary rat cortical neurons.
CONCLUSIONS
The study reveals that BMSC-sEVs protect rat cortical neurons from I/R injury which may be attributed to its anti-oxidant and anti-apoptosis activities via Ca2+/CaMK II pathway. Thus, BMSC-sEVs might be a therapeutic target for cerebral I/R injury after successful ROSC.
Funding: This work was supported by the Natural Science Foundation of China (81701872) and Medical Innovation Teams of Jiangsu Province (CXTDA2017007).
Ethical approval: All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC, Approval number: 1801008) of Nanjing Medical University.
Conflicts of interest: The authors indicated no potential conflicts of interest.
Contributors: SSG and XWK contributed equally to this study. All authors reviewed and approved the final version of manuscript for publication.
Reference
Categorization of survival and death after cardiac arrest
DOI:10.1016/j.resuscitation.2017.03.005
URL
PMID:28279695
[Cited within: 1]

BACKGROUND: Most cardiac arrest (CA) patients remain comatose post-resuscitation, prompting goals-of-care (GOC) conversations. The impact of these conversations on patient outcomes has not been well described. METHODS: Patients (n=385) treated for CA in Columbia University ICUs between 2008-2015 were retrospectively categorized into various modes of survival and death based on documented GOC discussions. Patients were deemed
Neurological prognostication of outcome in patients in coma after cardiac arrest
URL PMID:27017468 [Cited within: 1]
Benefits of using an endotracheal tube introducer as an adjunct to a Macintosh laryngoscope for endotracheal intubation performed by inexperienced doctors during mechanical CPR: A randomized prospective crossover study
DOI:10.5847/wjem.j.1920-8642.2019.03.009 URL PMID:31171950 [Cited within: 1]
Association between therapeutic hypothermia and survival after in-hospital cardiac arrest
URL PMID:27701659 [Cited within: 1]
Hypothermia and brain inflammation after cardiac arrest
DOI:10.4103/bc.bc_4_18
URL
PMID:30276330
[Cited within: 1]

The cessation (ischemia) and restoration (reperfusion) of cerebral blood flow after cardiac arrest (CA) induce inflammatory processes that can result in additional brain injury. Therapeutic hypothermia (TH) has been proven as a brain protective strategy after CA. In this article, the underlying pathophysiology of ischemia-reperfusion brain injury with emphasis on the role of inflammatory mechanisms is reviewed. Potential targets for immunomodulatory treatments and relevant effects of TH are also discussed. Further studies are needed to delineate the complex pathophysiology and interactions among different components of immune response after CA and identify appropriate targets for clinical investigations.
Clinical outcomes of transplanted modified bone marrow-derived mesenchymal stem cells in stroke: a phase 1/2a study
DOI:10.1161/STROKEAHA.116.012995
URL
PMID:27256670
[Cited within: 1]
BACKGROUND AND PURPOSE: Preclinical data suggest that cell-based therapies have the potential to improve stroke outcomes. METHODS: Eighteen patients with stable, chronic stroke were enrolled in a 2-year, open-label, single-arm study to evaluate the safety and clinical outcomes of surgical transplantation of modified bone marrow-derived mesenchymal stem cells (SB623). RESULTS: All patients in the safety population (N=18) experienced at least 1 treatment-emergent adverse event. Six patients experienced 6 serious treatment-emergent adverse events; 2 were probably or definitely related to surgical procedure; none were related to cell treatment. All serious treatment-emergent adverse events resolved without sequelae. There were no dose-limiting toxicities or deaths. Sixteen patients completed 12 months of follow-up at the time of this analysis. Significant improvement from baseline (mean) was reported for: (1) European Stroke Scale: mean increase 6.88 (95% confidence interval, 3.5-10.3; P<0.001), (2) National Institutes of Health Stroke Scale: mean decrease 2.00 (95% confidence interval, -2.7 to -1.3; P<0.001), (3) Fugl-Meyer total score: mean increase 19.20 (95% confidence interval, 11.4-27.0; P<0.001), and (4) Fugl-Meyer motor function total score: mean increase 11.40 (95% confidence interval, 4.6-18.2; P<0.001). No changes were observed in modified Rankin Scale. The area of magnetic resonance T2 fluid-attenuated inversion recovery signal change in the ipsilateral cortex 1 week after implantation significantly correlated with clinical improvement at 12 months (P<0.001 for European Stroke Scale). CONCLUSIONS: In this interim report, SB623 cells were safe and associated with improvement in clinical outcome end points at 12 months. CLINICAL TRIAL REGISTRATION: URL: https://www.clinicaltrials.gov. Unique identifier: NCT01287936.
Effect of adipose-derived mesenchymal stem cell administration and mild hypothermia induction on delayed neuronal death after transient global cerebral ischemia
DOI:10.1097/CCM.0000000000002289
URL
PMID:28252535
[Cited within: 1]
OBJECTIVES: Global cerebral ischemia is a cause of poor prognosis after resuscitation from cardiac arrest. Various attempts have been made to minimize global cerebral ischemia but none been more effective than mild hypothermia induction. A few studies have shown the effect of mesenchymal stem cells on global cerebral ischemia, but no studies have compared this effect with mild hypothermia or assessed any possible interaction. We aimed to show the effect of mesenchymal stem cells on delayed neuronal death after global cerebral ischemia and to compare this effect with mild hypothermia. DESIGN: Experimental study. SETTING: Animal research laboratory. SUBJECTS: Adult male Sprague-Dawley rats weighing 250-300 g. INTERVENTIONS: Rats were subjected to 7 minutes of transient global cerebral ischemia and randomized into four groups: control, mild hypothermia, injection of human adipose-derived mesenchymal stem cells, and combined application of mild hypothermia and mesenchymal stem cells, along with four sham groups treated identically. Rats were euthanized 7 days after global cerebral ischemia. MEASUREMENTS AND MAIN RESULTS: Degree of neuronal death in hippocampus was significantly higher in control than in other groups. The number of activated microglia was higher in control group than in other groups and was higher in mild hypothermia than shams, mesenchymal stem cells, mild hypothermia/mesenchymal stem cells. Degree of blood-brain barrier disruption and the count of infiltrated neutrophils were significantly higher in control than in other groups. Degree of oxidative injury was significantly higher in control than other groups. It was higher in mild hypothermia than sham groups, mesenchymal stem cells, mild hypothermia/mesenchymal stem cells and was higher in mesenchymal stem cells group than sham groups. Significantly, worse functional results were found in control than in other groups. CONCLUSIONS: Administration of mesenchymal stem cells after transient global cerebral ischemia has a prominent protective effect on delayed neuron death, even compared with mild hypothermia.
Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication
URL PMID:30602770 [Cited within: 2]
Exosomes derived from mesenchymal stem cells rescue myocardial ischaemia/reperfusion injury by inducing cardiomyocyte autophagy via AMPK and Akt pathways
DOI:10.1159/000480317
URL
PMID:28848091
[Cited within: 2]
BACKGROUND/AIMS: Reperfusion after an ischaemic insult might cause infarct extension. Mesenchymal stem cell (MSC)-derived exosomes could attenuate myocardial remodelling in animal models of myocardial ischaemia reperfusion injury (MIRI), and the present study aimed to explore the related mechanisms. METHODS: In vitro, rat H9C2 cardiomyocytes (H9C2s) were exposed to H2O2. Cell viability was detected by the CCK-8 assay, apoptosis was detected by Annexin V-PE/7-AAD staining, ROS production was detected by fluorescence microscopy and flow cytometry, and apoptosis-related proteins and signalling pathway-related proteins were detected by western blot analysis. Autophagic flux was measured using the tandem fluorescent mRFG-GFP-LC3 assay. MSC-derived exosomes were extracted using the total exosome isolation reagent. Apoptosis, myocardial infarction size, heart function and myocardial LC3B expression were examined in an in vivo I/R model by the TUNEL assay, TTC/Evan blue staining, echocardiography and immunohistochemicalstaining, respectively. RESULTS: In vitro, H2O2 dose-dependently increased ROS production and cell apoptosis in H9C2s and blocked autophagic flux after 3 h of exposure; autophagy gradually decreased thereafter, and the lowest level was detected at 12 h after exposure. MSC-derived exosomes reduced H2O2-induced ROS production and cell apoptosis and enhanced autophagy at 12 h after exposure. In H9C2 cells exposed to H2O2 for 12 h, treatment with exosomes enhanced autophagy via the AMPK/mTOR and Akt/mTOR pathways. Likewise, in vivo exosome injections in rats that underwent I/R injury significantly reduced apoptosis and the myocardial infarct size and upregulated myocardial LC3B expression as well as improved heart function. CONCLUSIONS: Our results indicate that MSC-derived exosomes could reduce MIRI by inducing cardiomyocyte autophagy via AMPK/mTOR and Akt/mTOR pathways.
Exosomes derived from mesenchymal stem cells ameliorate renal ischemic-reperfusion injury through inhibiting inflammation and cell apoptosis
Exosome-shuttled miR-92b-3p from ischemic preconditioned astrocytes protects neurons against oxygen and glucose deprivation
DOI:10.1016/j.brainres.2019.04.009
URL
PMID:30986407
[Cited within: 1]
Ischemic preconditioning (IPC) exerts protective effects against ischemic cerebral injury. In the present study, an in vitro model of cerebral ischemia (oxygen and glucose deprivation, OGD) was established to investigate the neuroprotective mechanism of IPC. We found that conditioned medium (C.M.) from astrocytes rather than neurons nor microglia cell line BV2 exerted neuroprotection. Moreover, exosomes derived from OGD preconditioned astrocytes can be taken up by neurons and attenuated OGD-induced neuron death and apoptosis. High-throughput microRNA (miRNA) sequencing revealed that miR-92b-3p levels in exosomes released from preconditioned astrocytes were increased. Overexpression of miR-92b-3p in neurons with miR-92b-3p mimic achieved the same protective effects as C.M. from astrocytes. Thus, we propose that the mechanism of IPC may associate with astrocytes, and that exosome-mediated miR-92b-3p shuttle from preconditioned astrocytes to neurons participate in these process.
Neuroprotective effects of pycnogenol against oxygen-glucose deprivation/reoxygenation-induced injury in primary rat astrocytes via NF-kappa B and ERK1/2 MAPK pathways
URL PMID:28662519 [Cited within: 1]
Mesenchymal stem cells secretome: a new paradigm for central nervous system regeneration?
URL PMID:23456256 [Cited within: 1]
Mesenchymal stem cell-derived extracellular vesicles: novel frontiers in regenerative medicine
DOI:10.1186/s13287-018-0791-7
URL
PMID:29523213
[Cited within: 1]
Mesenchymal stem cells (MSCs) are multipotent stem cells that have gained significant attention in the field of regenerative medicine. The differentiation potential along with paracrine properties of MSCs have made them a key option for tissue repair. The paracrine functions of MSCs are applied through secreting soluble factors and releasing extracellular vesicles like exosomes and microvesicles. Extracellular vesicles are predominantly endosomal in origin and contain a cargo of miRNA, mRNA, and proteins that are transferred from their original cells to target cells. Recently it has emerged that extracellular vesicles alone are responsible for the therapeutic effect of MSCs in plenty of animal diseases models. Hence, MSC-derived extracellular vesicles may be used as an alternative MSC-based therapy in regenerative medicine. In this review we discuss MSC-derived extracellular vesicles and their therapeutic potential in various diseases.
Mesenchymal stem cell-derived extracellular vesicles ameliorate hippocampal synaptic impairment after transient global ischemia
URL PMID:28769765 [Cited within: 1]
MicroRNA cluster miR-17-92 cluster in exosomes enhance neuroplasticity and functional recovery after stroke in rats
DOI:10.1161/STROKEAHA.116.015204
URL
PMID:28232590
[Cited within: 1]
BACKGROUND AND PURPOSE: Multipotent mesenchymal stromal cell (MSC) harvested exosomes are hypothesized as the major paracrine effectors of MSCs. In vitro, the miR-17-92 cluster promotes oligodendrogenesis, neurogenesis, and axonal outgrowth. We, therefore, investigated whether the miR-17-92 cluster-enriched exosomes harvested from MSCs transfected with an miR-17-92 cluster plasmid enhance neurological recovery compared with control MSC-derived exosomes. METHODS: Rats subjected to 2 hours of transient middle cerebral artery occlusion were intravenously administered miR-17-92 cluster-enriched exosomes, control MSC exosomes, or liposomes and were euthanized 28 days post-middle cerebral artery occlusion. Histochemistry, immunohistochemistry, and Golgi-Cox staining were used to assess dendritic, axonal, synaptic, and myelin remodeling. Expression of phosphatase and tensin homolog and activation of its downstream proteins, protein kinase B, mechanistic target of rapamycin, and glycogen synthase kinase 3beta in the peri-infarct region were measured by means of Western blots. RESULTS: Compared with the liposome treatment, both exosome treatment groups exhibited significant improvement of functional recovery, but miR-17-92 cluster-enriched exosome treatment had significantly more robust effects on improvement of neurological function and enhancements of oligodendrogenesis, neurogenesis, and neurite remodeling/neuronal dendrite plasticity in the ischemic boundary zone (IBZ) than the control MSC exosome treatment. Moreover, miR-17-92 cluster-enriched exosome treatment substantially inhibited phosphatase and tensin homolog, a validated miR-17-92 cluster target gene, and subsequently increased the phosphorylation of phosphatase and tensin homolog downstream proteins, protein kinase B, mechanistic target of rapamycin, and glycogen synthase kinase 3beta compared with control MSC exosome treatment. CONCLUSIONS: Our data suggest that treatment of stroke with tailored exosomes enriched with the miR-17-92 cluster increases neural plasticity and functional recovery after stroke, possibly via targeting phosphatase and tensin homolog to activate the PI3K/protein kinase B/mechanistic target of rapamycin/glycogen synthase kinase 3beta signaling pathway.
Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury
DOI:10.1186/s12974-018-1282-6
URL
PMID:30153825
[Cited within: 1]
BACKGROUND: Nod-like receptor protein 3 (NLRP3) inflammasome is a crucial factor in mediating inflammatory responses after cerebral ischemia/reperfusion (I/R), but the cellular location of NLRP3 inflammasome in cerebral I/R has yet come to a conclusion, and there is still no specific evidence to state the relationship between mitochondria and the NLRP3 inflammasome in cerebral I/R. METHODS: In the present study, we detected the cellular localization of NLRP3 inflammasomes in a transient middle cerebral artery occlusion (tMCAO) rat model and a transwell co-culture cell system under oxygen-glucose deprivation/reoxygenation (OGD/R) conditions. Then, we investigated the relationship between mitochondrial dysfunction and the activation of NLRP3 inflammasomes in different cell types after OGD/R and cerebral I/R injury. RESULTS: Our results showed that NLRP3 inflammasomes were first activated in microglia soon after cerebral I/R injury onset and then were expressed in neurons and microvascular endothelial cells later, but they were mainly in neurons. Furthermore, mitochondrial dysfunction played an important role in activating NLRP3 inflammasomes in microglia after OGD/R, and mitochondrial protector could inhibit the activation of NLRP3 inflammasomes in cerebral I/R rats. CONCLUSION: Our findings may provide novel insights into the cell type-dependent activation of NLRP3 inflammasomes at different stages of cerebral I/R injury and the role of mitochondrial dysfunction in activating the NLRP3 inflammasome pathway.
CaMK II as a pathological mediator of ER stress, oxidative stress, and mitochondrial dysfunction in a murine model of nephronophthisis
URL PMID:27076647 [Cited within: 1]
Tris (1,3-dichloro-2-propyl) phosphate induces toxicity by stimulating CaMK 2 in PC12 cells
DOI:10.1002/tox.22401
URL
PMID:28181390
[Cited within: 1]
Tris (1,3-dichloro-2-propyl) phosphate (TDCIPP) is one of the widely used organophosphorus flame retardants (OPFRs), which are regarded as suitable substitutes for brominated flame retardants (BFRs). Previously, we have validated the toxicity of TDCIPP in PC12 cells owing to the induced alterations in GAP43, NF-H, CaMK2a/2b, and tubulin alpha/beta proteins; however, limited information is currently available on the toxicity and mechanism of TDCIPP. In the present study, cytotoxicity effects were evaluated by exposing PC12 cells to different concentrations of TDCIPP (0-50 muM) for 4 days. To explore the possible mechanisms through which cytotoxicity is induced, changes in intracellular [Ca(2+) ]i levels and the activation of calmodulin dependent protein kinase 2 (CaMK2), c-Jun N-terminal kinase (JNK), extracellular regulated protein kinases (ERK1/2), and p38 mitogen-activated protein kinases (MAPK) pathways were evaluated. Furthermore, PC12 cells were pretreated with CaMK2 inhibitor KN93 to investigate the relationship between TDCIPP-induced phosphorylation of CaMK2 and activation of JNK, ERK1/2, and p38 MAPK pathways. Our results indicate that TDCIPP-induced toxicity might be associated with the overload of [Ca(2+) ]i levels, increased phosphorylation of CaMK2, and activation of the JNK, ERK1/2, and p38 MAPK pathways, the lattermost of which was further demonstrated to be partially elicited by the CaMK2 phosphorylation.
Calcium/calmodulin-dependent protein kinase II mediates group I metabotropic glutamate receptor-dependent protein synjournal and long-term depression in rat hippocampus
URL PMID:21593322 [Cited within: 1]
Multiple roles of calcium ions in the regulation of neurotransmitter release
DOI:10.1016/j.neuron.2008.08.019
URL
PMID:18817727
[Cited within: 1]
The intracellular calcium concentration ([Ca(2+)]) has important roles in the triggering of neurotransmitter release and the regulation of short-term plasticity (STP). Transmitter release is initiated by quite high concentrations within microdomains, while short-term facilitation is strongly influenced by the global buildup of
Calmodulin-kinases: modulators of neuronal development and plasticity
DOI:10.1016/j.neuron.2008.08.021
URL
[Cited within: 1]
In the nervous system, many intracellular responses to elevated calcium are mediated by CaM kinases (CaMKs), a family of protein kinases whose activities are initially modulated by binding Ca2+/calmodulin and subsequently by protein phosphorylation. One member of this family, CaMKII, is well-established for its effects on modulating synaptic plasticity and learning and memory. However, recent studies indicate that some actions on neuronal development and function attributed to CaMKII may instead or in addition be mediated by other members of the CaMK cascade, such as CaMKK, CaMKI, and CaMKIV. This review summarizes key neuronal functions of the CaMK cascade in signal transduction, gene transcription, synaptic development and plasticity, and behavior. The technical challenges of mapping cellular protein kinase signaling pathways are also discussed.
Simvastatin pretreatment protects cerebrum from neuronal injury by decreasing the expressions of phosphor-CaMK II and AQP4 in ischemic stroke rats
DOI:10.1007/s12031-014-0307-6
URL
PMID:24752488
[Cited within: 1]
Excitotoxicity and cytotoxic edema are the two major factors resulting in neuronal injury during brain ischemia and reperfusion. Ca2+/calmodulin-dependent protein kinase II (CaMK II), the downstream signal molecular of N-methyl-D-aspartate receptors (NMDARs), is a mediator in the excitotoxicity. Aquaporin 4 (AQP4), expressed mainly in the brain, is an important aquaporin to control the flux of water. In a previous study, we had reported that pretreatment of simvastatin protected the cerebrum from ischemia and reperfusion injury by decreasing neurological deficit score and infarct area (Zhu et al. PLoS One 7:e51552, 2012). The present study used a middle cerebral artery occlusion (MCAO) model to further explore the pleiotropic effect of simvastatin via CaMK II and AQP4. The results showed that simvastatin reduced degenerated cells and brain edema while decreasing the protein expressions of phosphor-CaMK II and AQP4, and increasing the ratios of Bcl-2/Bax, which was independent of cholesterol-lowering effect. Immunocomplexes formed between the subunit of NMDARs-NR3A and AQP4 were detected for the first time. It was concluded that simvastatin could protect the cerebrum from neuronal excitotoxicity and cytotoxic edema by downregulating the expressions of phosphor-CaMK II and AQP4, and that the interaction between NR3A and AQP4 might provide the base for AQP4 involving in the signaling pathways mediated by NMDARs.
Exosomes derived from miR-214-enriched bone marrow-derived mesenchymal stem cells regulate oxidative damage in cardiac stem cells by targeting CaMK II
DOI:10.1155/2018/4971261
URL
PMID:30159114
[Cited within: 1]
Cardiac stem cells (CSCs) have emerged as one of the most promising stem cells for cardiac protection. Recently, exosomes from bone marrow-derived mesenchymal stem cells (BMSCs) have been found to facilitate cell proliferation and survival by transporting various bioactive molecules, including microRNAs (miRs). In this study, we found that BMSC-derived exosomes (BMSC-exos) significantly decreased apoptosis rates and reactive oxygen species (ROS) production in CSCs after oxidative stress injury. Moreover, a stronger effect was induced by exosomes collected from BMSCs cultured under hypoxic conditions (Hypoxic-exos) than those collected from BMSCs cultured under normal conditions (Nor-exos). We also observed greater miR-214 enrichment in Hypoxic-exos than in Nor-exos. In addition, a miR-214 inhibitor or mimics added to modulate miR-214 levels in BMSC-exos revealed that exosomes from miR-214-depleted BMSCs partially reversed the effects of hypoxia-induced exosomes on oxidative damage in CSCs. These data further confirmed that miR-214 is the main effector molecule in BMSC-exos that protects CSCs from oxidative damage. miR-214 mimic and inhibitor transfection assays verified that CaMKII is a target gene of miR-214 in CSCs, with exosome-pretreated CSCs exhibiting increased miR-214 levels but decreased CaMKII levels. Therefore, the miR-214/CaMKII axis regulates oxidative stress-related injury in CSCs, such as apoptosis, calcium homeostasis disequilibrium, and excessive ROS accumulation. Collectively, these findings suggest that BMSCs release miR-214-containing exosomes to suppress oxidative stress injury in CSCs through CaMKII silencing.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}