


INTRODUCTION
Pulmonary fibrosis (PF) is a serious pulmonary interstitial disease.[1] The main pathological characteristics of PF are the proliferation of lung stromal cells and excessive deposition of extracellular matrix, which leads to the destruction of lung structure and function.[2] There are many causes of PF, including pathogenic microorganism infection, drug side effects, and toxicant exposure.[3,4] Some toxic poisons, such as paraquat (PQ), a bipyridine herbicide widely used around the world, accumulate in the lungs after ingestion, leading to lung injury and acute progressive fibrosis.[5,6] Although clinical experts have reached a consensus on the diagnosis and treatment of PQ poisoning, due to the absence of an effective antidote, the mortality rate of PQ poisoning remains as high as 50%-70%.[7⇓-9]
Previous studies have shown that approximately 30% of PF fibroblasts are of epithelial origin, and epithelial-mesenchymal transition (EMT) has been proven to be the core mechanism of PF.[10,11] The existing evidence shows that PQ can induce EMT phenotypes, which may be associated with oxidative injury after poisoning.[6,12] It is generally accepted that PQ causes the production of a significant amount of oxygen free radicals after being reduced to free radicals by a single electron with the aid of nicotinamide adenine dinucleotide phosphate (NADPH).[5⇓-7,13] As a highly dynamic organelle, excessive mitochondrial fission also plays a positive role in the oxidative stress response.[14] Nevertheless, the relationship between mitochondrial fission and oxidative stress in PQ-induced EMT remains unknown.
Pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) are mediators of inflammation in PF.[15] In noninfectious disease-related PF, inflammation is mainly evoked by DAMPs.[16] Mitochondria are not only the energy factories of cells but also the sources of DAMPs. Mitochondria-released DAMPs, particularly mitochondrial DNA (mtDNA), contribute to aseptic inflammation through the Toll-like receptor (TLR)-9 and nuclear factor kappa B (NF-κB) pathways.[16] Some studies have shown the mechanism of mtDNA efflux through the outer membrane of mitochondria.[17,18] In the process of mitochondrial fission, dynamin-related protein 1 (Drp1) forms a spiral oligomer, which wraps and splits the outer membrane of mitochondria.[19] Our previous study found that mitochondrial fission contributes to the release of cytochrome C, which indicates its potential role in the release of other DAMPs.
Using in vivo and in vitro PF models, this study investigated the relationship between mitochondrial fission and oxidative stress in PQ-induced EMT. Additionally, the effect of their crosstalk on the release of mtDNA and its mediating inflammatory response were explored.
METHODS
Animals
Healthy SPF male C57BL/6 mice, 6-8 weeks old, weighing 20-22 g, were purchased from Shanghai Vital River Laboratory Animal Technology Co., Ltd., China. All animal experiments were carried out according to the Guides for the Care and Use of Laboratory Animals, National Academy of Sciences, China. The present animal study was approved by the Laboratory Animal Ethics Committee of the First Affiliated Hospital of Wenzhou Medical University (WYYY-AEC-2022-0032).
In vivo experimental procedures
Experiment 1: The mice were randomly divided into two groups: the control group and the PQ group. At 12 h and days 1, 3 and 7 after exposure to PQ, lung tissue and blood samples were taken for further studies.
Experiment 2: Mitochondrial division inhibitor-1 (Mdivi-1), a specific mitochondrial fission inhibitor, was administered at 6 h after PQ exposure. The mice were randomly divided into four groups: control group, Mdivi-1 group, PQ group, and PQ+Mdivi-1 group.
Experiment 3: The mice were administered N-acetyl-L-cysteine (NAC), a classic antioxidant, at 6 h after PQ exposure. The mice were randomly divided into four groups: control group, NAC group, PQ group, and PQ+NAC group.
In vitro experimental procedures
MLE-12 cells (mouse lung type II epithelial cell line) were purchased from American Type Culture Collection (ATCC). The cells were cultured in DMEM-F12 (11320033, Thermo Fisher Scientific, USA) complete medium containing 10% fetal bovine serum and 1% antibiotics (penicillin and streptomycin). The cells were cultured at 37 ℃ and 5% CO2. When the density of MLE-12 cells on the plates reached 60%-70%, they were stimulated with PQ. Similar to in vivo studies, Mdivi-1 and NAC were used to inhibit mitochondrial fission and oxidative stress, respectively.
Hematoxylin and eosin (H&E) staining
The slices of left lung tissues were stained according to the instructions of the H&E staining kit (G1120, Beijing Solarbio Science & Technology Company Limited, China) and were viewed under an optical microscope. A semi-quantitative scoring system was introduced to evaluate lung injury, which included alveolar congestion, alveolar hemorrhage, neutrophil infiltration or aggregation in the alveolar cavity or vascular wall, alveolar wall thickening and/or transparent membrane formation. The four variables were summed to represent the lung injury score (total score: 0-16).
Western blotting
Total protein was extracted from MLE-12 cells and lung tissues. The primary antibodies used in this study were against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (ab8245, Abcam, USA), Drp-1 (8570S, Cell Signaling Technology, USA), E-cadherin (ab76055, Abcam, USA), α-smooth muscle actin (α-SMA) (19245S, Cell Signaling Technology, USA), TLR-9 (ab134368, Abcam, USA), phosphorylated (P)-NF-κB p65 (3033S, Cell Signaling Technology, USA), and NF-κB p65 (8242S, Cell Signaling Technology, USA).
Cell scratch test
MLE-12 cells (5,000 cells/well) were inoculated in 12-well plates and cultured at 37 °C and 5% CO2 to cover the bottom of the plate. Then, a straight line was drawn in the center of each well with a 100 μL sterile micropipette tip, and pictures were taken (0 h). Cells were treated with or without PQ and/or Mdivi-1. Then, 48 h later, the distance remaining was observed with an inverted microscope (Nikon, Japan) after cells of each group migrated from the scratch edge to the center.
Enzyme-linked immunosorbent assay (ELISA)
Mouse lung tissues were homogenized and centrifuge, and the supernatant was collected. The levels of interleukin 6 (IL-6), interleukin-1β (IL-1β), and tumor necrosis factor-α (TNF-α) in the tissue supernatant were detected by ELISA kits provided by Multi Sciences (EK206, EK201B, EK282HS, Multi Sciences, China).
Superoxide dismutase (SOD) activity and glutathione (GSH) and malondialdehyde (MDA) levels
In brief, mouse lung tissues were homogenized, and the homogenate was centrifuged. The supernatant was used to detect SOD activity and GSH and MDA levels according to the instructions (S0101, S0053, S0131, Beyotime Biotechnology, China).
Mitochondrial morphology
MLE-12 cells (5,000 cells/well) were inoculated in a 12-well plate with glass slides placed inside each well. The cells were cultured at 37 °C and 5% CO2 for 48 h. Then, the cells were stained with MitoTracker® Deep Red FM (200 nmol/L, M22426, Thermo Fisher Scientific, USA) and 6-diamidino-2-phenylindole (DAPI, 4083, Cell Signaling Technology, USA). The slides were viewed under a confocal laser scanning microscope (Nikon, Japan).
Real-time polymerase chain reaction (PCR) for mtDNA
Mouse blood samples were collected, and the plasma was extracted. The mtDNA in plasma and cell culture supernatant was extracted by the blood mtDNA extraction kit (XH007, Beijing Bio Lebo Technology Company Limited, China), and the levels of mtDNA were detected by the mouse mtDNA copy number assay kit (MCN3, Drtroit R&D, USA) based on real-time PCR.
Transmission electron microscopy (TEM)
The lung tissues of mice were fixed with 2.5% glutaraldehyde at 4 ℃ for 4 h and then fixed with 1% osmium tetroxide at 20 ℃ for 2 h. Then, the tissue was dehydrated by graded concentrations of ethanol and acetone, embedded in Epon812, sliced using an ultramicrotome, and stained with uranyl acetate and lead nitrate. The sections were observed by TEM (Hitachi, Japan).
Statistical analysis
Statistical analyses were performed using GraphPad 8.0 statistical program (GraphPad software, USA) and SPSS 22.0 statistical software (IBM, USA). The data are represented as the mean±standard deviation (SD). Student’s t-test was used to determine the differences between two groups, and the differences among multiple groups were determined by one-way analysis of variance (ANOVA) followed by least significant difference (LSD) post hoc analysis. The survival rate was analyzed by Kaplan-Meier analysis and compared by the log-rank test. A P-value <0.05 was considered as a statistically significant difference.
RESULTS
PQ caused EMT and PF in vivo and in vitro
The mice were treated with different concentrations of PQ, and 50 mg/kg PQ was used to induce the PF model (supplementary Figure 1A). H&E staining of lung sections showed that the acute lung injury score increased significantly at 12 h but decreased at 7 d after PQ exposure (supplementary Figure 1B). Gradually aggravated diffuse alveolar collapse and thickening suggest the occurrence of PF (supplementary Figure 1B). EMT in PQ-induced PF is evidenced by decreased levels of E-cadherin and increased expression of α-SMA in lung tissues (supplementary Figures 1 C-E). To examine whether PQ could induce EMT in vitro, we incubated MLE-12 cells with PQ for 12, 24 and 48 h. The results showed that PQ suppressed E-cadherin expression and up-regulated α-SMA expression in a time-dependent manner (supplementary Figures 1F-H). The cell scratch test showed decreased migration ability of MLE-12 cells after PQ stimulation, indicating the EMT of the cells (supplementary Figure 1I).
Mitochondrial fission and Drp1 expression increased in PQ-induced PF
At 24 h after PQ exposure, the lung had mitochondria with irregular shapes and disorganized cristae (supplementary Figure 2A). At 7 d after PQ exposure, mitochondrial morphology partially recovered (supplementary Figure 2A). In vitro, PQ-induced mitochondrial fragmentation and the content of mitochondria in MLE-12 cells were also reduced (supplementary Figure 2B). Drp1 is the main mediator of mitochondrial fission. We found that the expression of Drp1 in lung tissue significantly increased at 1 d and 3 d after PQ exposure but slightly decreased at 7 d, which was consistent with the morphological changes in mitochondria (supplementary Figures 2 C and E). In vitro, the peak expression of Drp1 appeared at 48 h after PQ stimulation (supplementary Figures 2 D and F).
Drp1-mediated mitochondrial fission alleviated EMT and PF
To determine the exact relationship between Drp1-mediated mitochondrial fission and EMT in PF, we treated mice with Mdivi-1 at 6 h post PQ exposure. One day later, Drp1 expression and mitochondrial fission were significantly decreased in the lung issues of mice treated with Mdivi-1 compared with those in the control group (Figures 1 A-C). Similarly, in MLE-12 cells exposed to PQ, Mdivi-1 treatment also downregulated the expression of Drp1, reduced mitochondrial fission, and increased mitochondrial content (Figures 1D-F).
Figure 1.
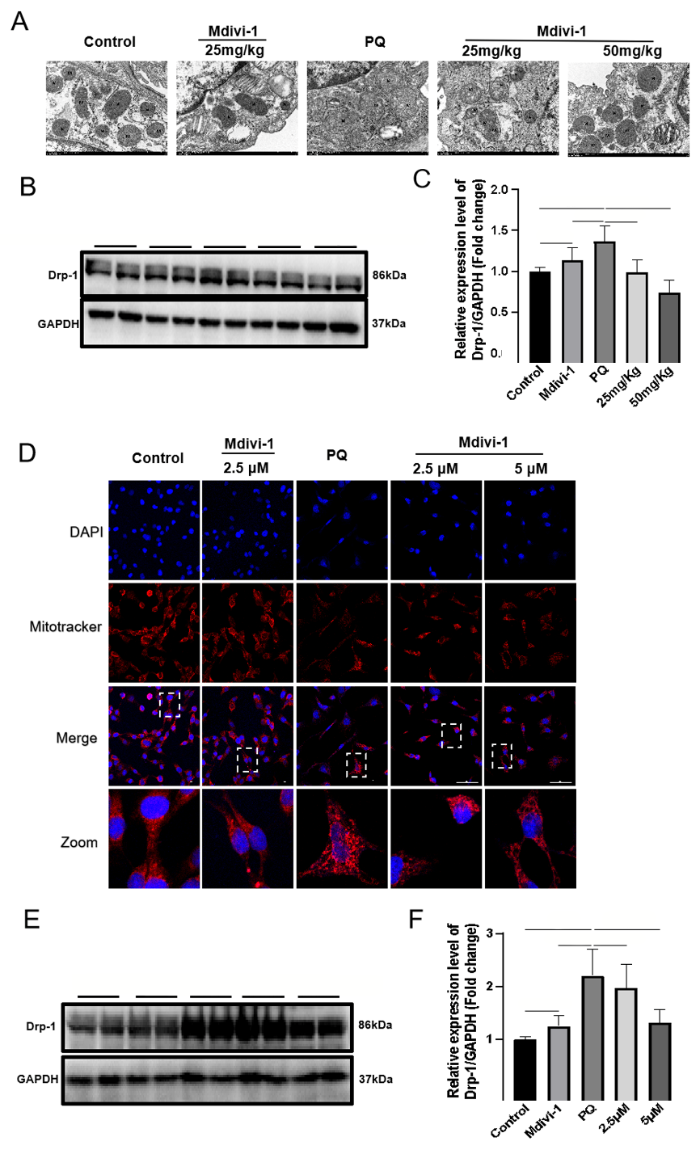
Figure 1.
Effects of Midivi-1 treatment on mitochondrial morphology and Drp1 expression in PQ-induced PF. A: morphological changes in mitochondria in lung tissues observed by transmission electron microscopy (original magnification, ×200); B, C: representative immunoblots and quantitative histogram of Drp1 in lung tissues from mice (n=6); D: MitoTracker® Deep Red FM and DAPI used to stain mitochondria and nuclei, and mitochondrial morphology determined using confocal laser microscopy (original magnification, ×200); E, F: representative immunoblots and quantitative histogram of Drp1 in MLE-12 cells. *P<0.05, **P<0.01. All values are denoted as the mean±SD. PQ: paraquat; Drp-1: dynamin-related protein 1; Midivi-1: mitochondrial division inhibitor-1; PF: pulmonary fibrosis; 25 mg/kg: PQ+Mdivi-1 (25 mg/kg); 50 mg/kg: PQ+Mdivi-1 (50 mg/kg); 2.5 μmol/L: PQ+Mdivi-1 (2.5 μmol/L); 5.0 μmol/L: PQ+Mdivi-1 (5.0 μmol/L); GAPDH: glyceraldehyde-3-phosphate dehydrogenase; ns: not significant.
The decrease in diffuse alveolar collapse and thickening indicates the role of Drp1-mediated mitochondrial fission in PF (supplementary Figures 3 A-B), which is confirmed by the decrease in α-SMA levels and increased E-cadherin expression after Mdivi-1 treatment (supplementary Figures 3 C-E). In MLE-12 cells, treatment with Mdivi-1 at 6 h after PQ stimulation significantly enhanced cell migration (supplementary Figure 4A), accompanied by a decrease in α-SMA expression and an increase in E-cadherin expression (supplementary Figures 4 B-D). Taken together, these results supported that Drp1-mediated mitochondrial fission contributed to EMT in PQ-induced PF.
We found that PQ induced increased oxidative stress, as evidenced by elevated MDA production and decreased GSH levels and SOD activity (supplementary Figures 4 E-G). Inhibiting Drp1-mediated mitochondrial fission by Mdivi-1 administration dramatically reduced oxidative stress injury as determined by increases in SOD activity and the levels of GSH and by decreases in MDA production (supplementary Figures 4 E-G).
NAC downregulated Drp1 expression and decreased mitochondrial fission and EMT
NAC administration was associated with a decrease in MDA levels and increases in SOD activity and GSH levels (supplementary Figures 5 A-C). NAC administration attenuated mitochondrial fission, which was determined by the reduction in Drp1 expression and mitochondrial structural damage (supplementary Figures 5 D-F). Compared with the control group, NAC administration decreased EMT, as evidenced by decreased α-SMA expression and increased E-cadherin levels (supplementary Figures 5 G-I). In PF mice, histological analysis showed decreased diffuse alveolar collapse and thickening after treatment with NAC (supplementary Figures 5 J and K).
mtDNA-mediated inflammation decreased after inhibiting Drp1-mediated mitochondrial fission
The over-expression of cytokines and subsequent inflammatory damage are important mechanisms of EMT.[20] In PQ-induced PF, the levels of IL-6, IL-1β and TNF-α in plasma were significantly increased, which was reversed by a Drp1 inhibitor (Figures 2 A-C). The levels of mtDNA, TLR9 expression, and P-NF-κB P65 were significantly increased in PQ-induced PF (Figures 2 E-G). Inhibiting Drp1-mediated mitochondrial fission with a Drp1 inhibitor reduced mtDNA release, as determined by a decrease in mtDNA levels in plasma and decreases in TLR9 and P-NF-κB P65 expression (Figures 2 E-G), but the Drp1 inhibitor did not affect the level of total P65. Together, these results demonstrated that targeting Drp1-mediated mitochondrial fission prevented mtDNA release and its mediated inflammation.
Figure 2.
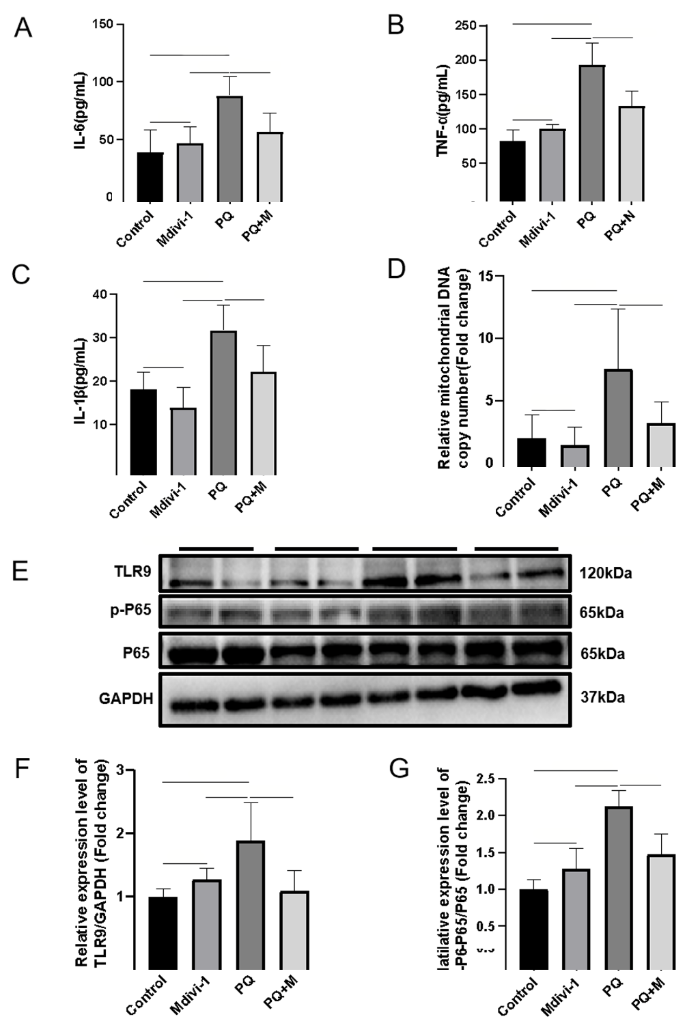
Figure 2.
mtDNA-mediated inflammation decreased after inhibiting Drp1-mediated mitochondrial fission. A-C: concentrations of IL-6, TNF-α, and IL-1β from mice measured by ELISA; D: the relative levels of mtDNA in plasma from mice; E-G: representative immunoblots and quantitative histogram of TLR9 and p-P65/P65 in the lung tissues from mice. n=6; *P<0.05, **P<0.01. All values are expressed as the mean±standard deviation. mtDNA: mitochondrial DNA; PQ: paraquat; TLR9: Toll-like receptor 9; P65: NF-κB p65; p-65: (P)-NF-κB p65; GAPDH: glyceraldehyde-3-phosphate dehydrogenase;ns: not significant.
NAC suppressed mtDNA release and activation of the TLR9-NF-κB signaling pathway
We determined whether oxidative stress contributes to mtDNA release. Mice receiving NAC at 6 h after PQ exposure had lower levels of mtDNA in the plasma, indicating that oxidative stress was crucial to the release of mtDNA in PF (supplementary Figure 6A). NAC treatment also reduced lung inflammation, as evidenced by decreased levels of cytokines, including IL-6, IL-1β and TNF-α (supplementary Figures 6 B-D). Additionally, the levels of TLR9 and P-NF-κB P65 in the lung also decreased after NAC treatment (supplementary Figures 6 E-G). Thus, our results demonstrated that oxidative stress promoted mtDNA-mediated inflammation in PF.
DISCUSSION
Mitochondria are dynamic organelles that undergo fusion and fission to maintain their function and morphology. Currently, an imbalance in mitochondrial fusion and fission has been proven to be involved in the pathogenesis of sepsis.[21] The role of mitochondrial fusion and fission in PF is not completely known. Here, we prove that increased mitochondrial fission significantly induces EMT in the PQ-induced PF mice model. Mechanistically, increased mitochondrial fission contributes to oxidative damage and the activation of the mtDNA/TLR9/NF-κB signaling pathway, which are the two main mechanisms of EMT.[11,22]
Oxidative stress is responsible for PQ-induced lung injury. Previous studies have also illustrated the crucial role of oxidative stress in EMT.[11,22] In diabetic nephropathy, targeted ROS alleviated EMT and renal fibrosis.[23] Chen et al[24] found that fangchinoline decreased ROS production and reversed EMT in non-small cell lung cancer, inhibiting cell invasion and migration. In this study, during PQ-induced EMT, we found that treatment with NAC, a ROS scavenger, not only significantly reduced oxidative damage and EMT but also attenuated mitochondrial fission and Drp1 expression, suggesting that Drp1-mediated mitochondrial fission is involved in oxidative-stress-induced EMT. Mitochondria are one of the sources of ROS in mammalian cells.[13] We found that the inhibition of Drp1-dependent mitochondrial fission by Mdivi-1, indicating increased generation of ROS, requires mitochondrial fission in PQ-induced EMT. Together, the present study showed that mutual promotion of mitochondrial fission and oxidative stress contributes to EMT in PQ-induced PF. Nevertheless, some studies have also shown that a lack of Drp1 leads to mitochondrial elongation and cumulative oxidative damage.[25] Thus, when targeting Drp1 to treat related diseases, the balance of mitochondrial fission and fusion should be fully evaluated.
Blocking TLR9 has also been shown to reduce bleomycin-induced lung injury.[26] To date, oxidative stress and TLR9-mediated inflammation are often used to explain the development of EMT and PF, although one study showed that the ROS inhibitor apocynin could attenuate ox-LDL-mediated mtDNA damage and TLR9 expression.[27] mtDNA shares unmethylated CpG DNA repeats with bacterial DNA and is a ligand for TLR9.[28] We found that treatment with NAC significantly reduced mtDNA release, TLR9 expression and the activation of NF-κB p65. Interestingly, suppression of Drp1-mediated mitochondrial fission also alleviated mtDNA release in vivo and in vitro. Similarly, Zhang et al[29] found that excessive mitochondrial fission was one of the reasons for mtDNA release in LPS-treated Kupffer cells. As mitochondrial fission and Drp1 expression were reduced after NAC treatment in the present study, oxidative stress may contribute to mtDNA release and inflammation through Drp1-mediated excessive mitochondrial fission.
Recently, many studies have reported the mechanism of mtDNA release. In the process of cell apoptosis, Bcl-2-associated X protein (BAX) and Bcl2 antagonist/killer (BAK) oligomerize to form pores in the outer membrane of mitochondria, which mediates the release of mtDNA.[17] In addition, under oxidative stress conditions, mitochondria release short mtDNA fragments through the pores formed by voltage-dependent anion channel (VDAC) oligomers.[18] Increased levels of Drp1 and its interaction with Bax/Bak and VDAC lead to the formation of pores that mediate mtDNA release.[30] Although the mechanisms underlying mtDNA release need to be further ruled out, targeting Bax/Bak or VDAC1 might be a potential treatment strategy for PQ-induced inflammation.
CONCLUSION
Mutual promotion of mitochondrial fission and oxidative stress contributes to EMT in PQ-induced PF, which is associated with the mtDNA/TLR9/NF-κB pathway.
Funding: The study was supported by the Wenzhou Municipal Science and Technology Bureau (Y2020092) and partly by the Key Specialty of Traditional Chinese Medicine of Zhejiang Province in the 13th Five-Year Plan period (Emergency Department).
Ethical approval: The study was approved by the Laboratory Animal Ethics Committee of Wenzhou Medical University (WYYY-AEC-2022-0032).
Conflicts of interest: The authors declare that there are no conflicts of interest regarding the publication of this paper.
Contributors: ZQL conceptualized and drafted the manuscript. JZ, WJL, SQC, and CZ performed the experiments. GJZ and ZQL designed the experiments and supervised and revised the manuscript. All authors made an individual contribution to the final approval of the version published.
All the supplementary files in this paper are available at http://wjem.com.cn.
Reference
Role of epithelial-to-mesenchymal transition in the pulmonary fibrosis induced by paraquat in rats
DOI:10.5847/wjem.j.1920-8642.2021.03.009 URL [Cited within: 1]
Targeting fibrosis, mechanisms and cilinical trials
Cellular and molecular mechanisms of fibrosis
DOI:10.1002/path.2277
PMID:18161745
[Cited within: 1]

Fibrosis is defined by the overgrowth, hardening, and/or scarring of various tissues and is attributed to excess deposition of extracellular matrix components including collagen. Fibrosis is the end result of chronic inflammatory reactions induced by a variety of stimuli including persistent infections, autoimmune reactions, allergic responses, chemical insults, radiation, and tissue injury. Although current treatments for fibrotic diseases such as idiopathic pulmonary fibrosis, liver cirrhosis, systemic sclerosis, progressive kidney disease, and cardiovascular fibrosis typically target the inflammatory response, there is accumulating evidence that the mechanisms driving fibrogenesis are distinct from those regulating inflammation. In fact, some studies have suggested that ongoing inflammation is needed to reverse established and progressive fibrosis. The key cellular mediator of fibrosis is the myofibroblast, which when activated serves as the primary collagen-producing cell. Myofibroblasts are generated from a variety of sources including resident mesenchymal cells, epithelial and endothelial cells in processes termed epithelial/endothelial-mesenchymal (EMT/EndMT) transition, as well as from circulating fibroblast-like cells called fibrocytes that are derived from bone-marrow stem cells. Myofibroblasts are activated by a variety of mechanisms, including paracrine signals derived from lymphocytes and macrophages, autocrine factors secreted by myofibroblasts, and pathogen-associated molecular patterns (PAMPS) produced by pathogenic organisms that interact with pattern recognition receptors (i.e. TLRs) on fibroblasts. Cytokines (IL-13, IL-21, TGF-beta1), chemokines (MCP-1, MIP-1beta), angiogenic factors (VEGF), growth factors (PDGF), peroxisome proliferator-activated receptors (PPARs), acute phase proteins (SAP), caspases, and components of the renin-angiotensin-aldosterone system (ANG II) have been identified as important regulators of fibrosis and are being investigated as potential targets of antifibrotic drugs. This review explores our current understanding of the cellular and molecular mechanisms of fibrogenesis.2007 Pathological Society of Great Britain and Ireland
Editorial: Pulmonary fibrosis: one manifestation, various diseases
DOI:10.3389/fphar.2022.1027332 URL [Cited within: 1]
Advances in the mechanism of paraquat-induced pulmonary injury
Performance of extracorporeal membrane oxygenation in patients with fatal paraquat poisoning: grasp for straws
DOI:10.5847/wjem.j.1920-8642.2021.03.013 URL [Cited within: 4]
An expert consensus on diagnosis and treatment of acute paraquat poisoning (2022)
Early metabolome profiling and prognostic value in paraquat-poisoned patients: based on ultraperformance liquid chromatography coupled to quadrupole time-of-flight mass spectrometry
DOI:10.1021/acs.chemrestox.7b00240
PMID:29099997
[Cited within: 1]
Paraquat (PQ) has caused countless deaths throughout the world. There remains no effective treatment for PQ poisoning. Here we study the blood metabolome of PQ-poisoned patients using ultraperformance liquid chromatography coupled to quadrupole time-of-flight mass spectrometry (UPLC/Q-TOF MS). Patients were divided into groups according to blood PQ concentration. Healthy subjects served as controls. Metabolic features were statistically analyzed using multivariate pattern-recognition techniques to identify the most important metabolites. Selected metabolites were further compared with a series of clinical indexes to assess the prognostic value. PQ-poisoned patients showed substantial differences compared with healthy subjects. Based on variable of importance in the project (VIP) values and statistical analysis, 17 metabolites were selected and identified. These metabolites well-classified low PQ-poisoned patients, high PQ-poisoned patients, and healthy subjects, which was better than that of a complete blood count (CBC). Among the 17 metabolites, 20:3/18:1-PC (PC), LPA (0:0/16:0) (LPA), 19-oxo-deoxycorticosterone (19-oxo-DOC), and eicosapentaenoic acid (EPA) had prognostic value. In particular, EPA was the most sensitive one. Besides, the levels of EPA was correlated with LPA and 19-oxo-DOC. If EPA was excessively consumed, then prognosis was poor. In conclusion, the serum metabolome is substantially perturbed by PQ poisoning. EPA is the most important biomarker in early PQ poisoning.
An effective machine learning approach for prognosis of paraquat poisoning patients using blood routine indexes
DOI:10.1111/bcpt.2017.120.issue-1 URL [Cited within: 1]
Pathogenesis of idiopathic pulmonary fibrosis
DOI:10.1146/annurev-pathol-012513-104706
PMID:24050627
[Cited within: 1]
Idiopathic pulmonary fibrosis (IPF) is a fibrosing interstitial lung disease associated with aging that is characterized by the histopathological pattern of usual interstitial pneumonia. Although an understanding of the pathogenesis of IPF is incomplete, recent advances delineating specific clinical and pathologic features of IPF have led to better definition of the molecular pathways that are pathologically activated in the disease. In this review we highlight several of these advances, with a focus on genetic predisposition to IPF and how genetic changes, which occur primarily in epithelial cells, lead to activation of profibrotic pathways in epithelial cells. We then discuss the pathologic changes within IPF fibroblasts and the extracellular matrix, and we conclude with a summary of how these profibrotic pathways may be interrelated.
Epithelial-mesenchymal transition in the pathogenesis of idiopathic pulmonary fibrosis
Epigenetic regulation of interleukin 6 by histone acetylation in macrophages and its role in paraquat-induced pulmonary fibrosis
Crosstalk between mitochondrial fission and oxidative stress in paraquat-induced apoptosis in mouse alveolar type ii cells
DOI:10.7150/ijbs.18468
PMID:28808421
[Cited within: 2]
Paraquat (PQ), as a highly effective and nonselective herbicide, induces cell apoptosis through generation of superoxide anions which forms reactive oxygen species (ROS). Mitochondria, as regulators for cellular redox signaling, have been proved to play an important role in PQ-induced cell apoptosis. This study aimed to evaluate whether and how mitochondrial fission interacts with oxidative stress in PQ-induced apoptosis in mouse alveolar type II (AT-II) cells. Firstly, we demonstrated that PQ promoted apoptosis and release of cytochrome-c (Cyt-c). Furthermore, we showed that PQ broke down mitochondrial network, enhanced the expression of fission-related proteins, increased Drp1 mitochondrial translocation while decreased the expression of fusion-related proteins in AT-II cells. Besides, inhibiting mitochondrial fission using mdivi-1, a selective inhibitor of Drp1, markedly attenuated PQ-induced apoptosis, release of Cyt-c and the generation of ROS. These results indicate that mitochondrial fission involves in PQ-induced apoptosis. Further study demonstrated that antioxidant ascorbic acid inhibited Drp1 mitochondrial translocation, mitochondrial fission and attenuated PQ-induced apoptosis. Overall, our findings suggest that mitochondrial fission interplays with ROS in PQ-induced apoptosis in mouse AT-II cells and mitochondrial fission could serve as a potential therapeutic target in PQ poisoning.
Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases
Circulating mitochondrial DAMPs cause inflammatory responses to injury
DOI:10.1038/nature08780 [Cited within: 1]
Mitochondrial control of inflammation
SFTSV infection induces BAK/BAX-dependent mitochondrial DNA release to trigger NLRP 3 inflammasome activation
DOI:10.1016/j.celrep.2020.02.105 URL [Cited within: 2]
VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease
DOI:10.1126/science.aav4011
PMID:31857488
[Cited within: 2]
Mitochondrial stress releases mitochondrial DNA (mtDNA) into the cytosol, thereby triggering the type Ι interferon (IFN) response. Mitochondrial outer membrane permeabilization, which is required for mtDNA release, has been extensively studied in apoptotic cells, but little is known about its role in live cells. We found that oxidatively stressed mitochondria release short mtDNA fragments via pores formed by the voltage-dependent anion channel (VDAC) oligomers in the mitochondrial outer membrane. Furthermore, the positively charged residues in the N-terminal domain of VDAC1 interact with mtDNA, promoting VDAC1 oligomerization. The VDAC oligomerization inhibitor VBIT-4 decreases mtDNA release, IFN signaling, neutrophil extracellular traps, and disease severity in a mouse model of systemic lupus erythematosus. Thus, inhibiting VDAC oligomerization is a potential therapeutic approach for diseases associated with mtDNA release.Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Vesicle transport related protein Synaptotagmin-1 mediates paraquat transport to antagonize paraquat toxicity
DOI:10.1016/j.tox.2022.153180 URL [Cited within: 2]
Mifepristone-inducible recombinant adenovirus attenuates paraquat-induced lung injury in rats
DOI:10.1177/0960327114532381
PMID:24812154
[Cited within: 1]
To investigate the effects of overexpression of nuclear factor E2-related factor-2 (NRF2) on lung injury in rats exposed to paraquat (PQ) poisoning.A mifepristone (RU486)-inducible recombinant adenoviral vector carrying the human NRF2 gene (Ad-RUNRF2) was constructed and transfected via airway into the rats 7 days before the administration of RU486. Rats were orally challenged with PQ at 20 mg/kg 24 h after the injection of RU486. On days 0.5, 3 and 21 after PQ poisoning, the expressions of NRF2 and cytokines related to inflammation and oxidation in lung tissue were examined.RU486 remarkably enhanced NRF2 mRNA and NRF2 protein levels in Ad-RUNRF2-transfected rats in a dose-dependent manner (p < 0.01). PQ stimulated compensatory overexpression of NRF2, heme oxygenase 1 (HO-1) and NAD(P)H quinone oxidoreductase 1 (NQO-1) in lungs on days 0.5 and 3 after exposure (p < 0.05), but depleted the expression of catalase (CAT), glutathione peroxidase (GSH-Px) and glutathione (GSH), with an increased malondialdehyde (MDA) (p < 0.05). However, pretreatment with Ad-RUNRF2 and RU486 strongly enhanced the expression levels of NRF2, HO-1, NQO-1, CAT and GSH-Px in the lungs of PQ intoxicated rats, with increased GSH and decreased MDA (p < 0.05). Pretreatment with Ad-RUNRF2 and RU486 also strongly suppressed the PQ-induced activation of nuclear factor κB (NF-κB) and decreased the levels of tumour necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and interleukin-6 (IL-6). In addition, Ad-RUNRF2 and RU486 induction significantly reduced PQ-induced pathological changes in lungs and attenuated lung oedema and protein leakage caused by PQ (p < 0.05).RU486-induced overexpression of NRF2 in lungs transfected with Ad-RUNRF2 can ameliorate PQ-induced lung injury by the activation of the NRF2-antioxidant response element (ARE) pathway.© The Author(s) 2014.
Mitochondrial quality control mechanisms as potential therapeutic targets in sepsis-induced multiple organ failure
DOI:10.1007/s00109-019-01756-2
PMID:30788535
[Cited within: 1]
Sepsis is a dysregulated response to severe infection characterized by life-threatening organ failure and is the leading cause of mortality worldwide. Multiple organ failure is the central characteristic of sepsis and is associated with poor outcome of septic patients. Ultrastructural damage to the mitochondria and mitochondrial dysfunction are reported in sepsis. Mitochondrial dysfunction with subsequent ATP deficiency, excessive reactive oxygen species (ROS) release, and cytochrome c release are all considered to contribute to organ failure. Consistent mitochondrial dysfunction leads to reduced mitochondrial quality control capacity, which eliminates dysfunctional and superfluous mitochondria to maintain mitochondrial homeostasis. Mitochondrial quality is controlled through a series of processes including mitochondrial biogenesis, mitochondrial dynamics, mitophagy, and transport processes. Several studies have indicated that multiple organ failure is ameliorated by restoring mitochondrial quality control mechanisms and is further amplified by defective quality control mechanisms. This review will focus on advances concerning potential mechanisms in regulating mitochondrial quality control and impacts of mitochondrial quality control on the progression of sepsis.
Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis
DOI:10.1007/s00018-020-03693-7
PMID:33201251
[Cited within: 2]
Idiopathic pulmonary fibrosis (IPF), the most common form of idiopathic interstitial pneumonia, is a progressive, irreversible, and typically lethal disease characterized by an abnormal fibrotic response involving vast areas of the lungs. Given the poor knowledge of the mechanisms underpinning IPF onset and progression, a better understanding of the cellular processes and molecular pathways involved is essential for the development of effective therapies, currently lacking. Besides a number of established IPF-associated risk factors, such as cigarette smoking, environmental factors, comorbidities, and viral infections, several other processes have been linked with this devastating disease. Apoptosis, senescence, epithelial-mesenchymal transition, endothelial-mesenchymal transition, and epithelial cell migration have been shown to play a key role in IPF-associated tissue remodeling. Moreover, molecules, such as chemokines, cytokines, growth factors, adenosine, glycosaminoglycans, non-coding RNAs, and cellular processes including oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress, hypoxia, and alternative polyadenylation have been linked with IPF development. Importantly, strategies targeting these processes have been investigated to modulate abnormal cellular phenotypes and maintain tissue homeostasis in the lung. This review provides an update regarding the emerging cellular and molecular mechanisms involved in the onset and progression of IPF.
Clinical efficacies, underlying mechanisms and molecular targets of Chinese medicines for diabetic nephropathy treatment and management
DOI:10.1016/j.apsb.2020.12.020
PMID:34589395
[Cited within: 1]
Diabetic nephropathy (DN) has been recognized as a severe complication of diabetes mellitus and a dominant pathogeny of end-stage kidney disease, which causes serious health problems and great financial burden to human society worldwide. Conventional strategies, such as renin-angiotensin-aldosterone system blockade, blood glucose level control, and bodyweight reduction, may not achieve satisfactory outcomes in many clinical practices for DN management. Notably, due to the multi-target function, Chinese medicine possesses promising clinical benefits as primary or alternative therapies for DN treatment. Increasing studies have emphasized identifying bioactive compounds and molecular mechanisms of reno-protective effects of Chinese medicines. Signaling pathways involved in glucose/lipid metabolism regulation, antioxidation, anti-inflammation, anti-fibrosis, and podocyte protection have been identified as crucial mechanisms of action. Herein, we summarize the clinical efficacies of Chinese medicines and their bioactive components in treating and managing DN after reviewing the results demonstrated in clinical trials, systematic reviews, and meta-analyses, with a thorough discussion on the relative underlying mechanisms and molecular targets reported in animal and cellular experiments. We aim to provide comprehensive insights into the protective effects of Chinese medicines against DN.© 2021 Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences. Production and hosting by Elsevier B.V.
Fangchinoline inhibits non-small cell lung cancer metastasis by reversing epithelial-mesenchymal transition and suppressing the cytosolic ROS-related Akt-mTOR signaling pathway
DOI:10.1016/j.canlet.2022.215783 URL [Cited within: 1]
Dynamin-related protein 1 deficiency leads to receptor-interacting protein kinase 3-mediated necroptotic neurodegeneration
DOI:S0002-9440(16)30308-X
PMID:27640145
[Cited within: 1]
Mitochondria are dynamic organelles that divide and fuse to modulate their number and shape. We have previously reported that the loss of dynamin-related protein 1 (Drp1), which mediates mitochondrial division, leads to the degeneration of cerebellar Purkinje cells in mice. Because Drp1 has been shown to be important for apoptosis and necroptosis, it is puzzling how Purkinje neurons die in the absence of Drp1. In this study, we tested whether neurodegeneration involves necrotic cell death by generating Purkinje cell-specific Drp1-knockout (KO) mice that lack the receptor-interacting protein kinase 3 (Rip3), which regulates necroptosis. We found that the loss of Rip3 significantly delays the degeneration of Drp1-KO Purkinje neurons. In addition, before neurodegeneration, mitochondrial tubules elongate because of unopposed fusion and subsequently become large spheres as a result of oxidative damage. Surprisingly, Rip3 loss also helps Drp1-KO Purkinje cells maintain the elongated morphology of the mitochondrial tubules. These data suggest that Rip3 plays a role in neurodegeneration and mitochondrial morphology in the absence of mitochondrial division.Copyright © 2016 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Blocking Toll-like receptor 9 attenuates bleomycin-induced pulmonary injury
DOI:10.4132/jptm.2021.12.27
PMID:35220710
[Cited within: 1]
Acute respiratory distress syndrome (ARDS) is one of the most common complications in coronavirus disease 2019 patients suffering from acute lung injury (ALI). In ARDS, marked distortion of pulmonary architecture has been reported. The pulmonary lesions in ARDS include hemodynamic derangements (such as alveolar edema and hemorrhage), vascular and bronchiolar damage, interstitial inflammatory cellular aggregations, and eventually fibrosis. Bleomycin induces ARDS-representative pulmonary damage in mice and rats; therefore, we used bleomycin model mice in our study. Recently, Toll-like receptor 9 (TLR9) was implicated in the development of ARDS and ALI.In this study, we evaluated the efficiency of a TLR9 blocker (ODN2088) on bleomycin-induced pulmonary damage. We measured the apoptosis rate, inflammatory reaction, and fibroplasia in bleomycin- and bleomycin + ODN2088-treated mice.Our results showed a significant amelioration in bleomycin-induced damage to pulmonary architecture following ODN2088 treatment. A marked decrease in pulmonary epithelial and endothelial apoptosis rate as measured by cleaved caspase-3 expression, inflammatory reaction as indicated by tumor necrosis factor α expression, and pulmonary fibrosis as demonstrated by Van Gieson staining and α-smooth muscle actin immunohistochemistry were observed following ODN2088 treatment.All these findings indicate that blocking downstream TLR9 signaling could be beneficial in prevention or mitigation of ARDS through hemodynamic derangements, inflammation, apoptosis, and fibrosis.
Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis
DOI:10.1038/srep01077
PMID:23326634
[Cited within: 1]
Our studies in HUVECs show that ox-LDL induced autophagy and damaged mtDNA leading to TLR9 expression. LOX-1 antibody or the ROS inhibitor apocynin attenuated ox-LDL-mediated autophagy, mtDNA damage and TLR9 expression, suggesting that these events are LOX-1 and ROS-dependent phenomena. Experiments using siRNA to DNase II indicated that DNase II digests mtDNA to protect the tissue from inflammation. Next, we studied and found intense autophagy, TLR9 expression and inflammatory signals (CD45 and CD68) in the aortas of LDLR knockout mice fed high cholesterol diet. Deletion of LOX-1 (LDLR/LOX-1 double knockout mice) attenuated autophagy, TLR9 expression as well as CD45 and CD68. Damaged mtDNA signal was also very high in LDLR knockout mice aortas, and this signal was attenuated by LOX-1 deletion. Thus, it appears that oxidative stress-mediated damaged mtDNA that escapes autophagy induces a potent inflammatory response in atherosclerosis.
Mitochondrial DNA in the regulation of innate immune responses
DOI:10.1007/s13238-015-0222-9
[Cited within: 1]
<p>Mitochondrion is known as the energy factory of the cell, which is also a unique mammalian organelle and considered to be evolved from aerobic prokaryotes more than a billion years ago. Mitochondrial DNA, similar to that of its bacterial ancestor’s, consists of a circular loop and contains significant number of unmethylated DNA as CpG islands. The innate immune system plays an important role in the mammalian immune response. Recent research has demonstrated that mitochondrial DNA (mtDNA) activates several innate immune pathways involving TLR9, NLRP3 and STING signaling, which contributes to the signaling platforms and results in effector responses. In addition to facilitating antibacterial immunity and regulating antiviral signaling, mounting evidence suggests that mtDNA contributes to inflammatory diseases following cellular damage and stress. Therefore, in addition to its well-appreciated roles in cellular metabolism and energy production, mtDNA appears to function as a key member in the innate immune system. Here, we highlight the emerging roles of mtDNA in innate immunity.</p>
STING signaling sensing of DRP1-dependent mtDNA release in kupffer cells contributes to lipopolysaccharide-induced liver injury in mice
DOI:10.1016/j.redox.2022.102367 URL [Cited within: 1]
DRP1 interacts directly with BAX to induce its activation and apoptosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}