


INTRODUCTION
Paraquat (PQ, 1,1’-dimethyl-4,4’-bipyridylium), also known as methyl viologen, has been widely used as a broad-spectrum, highly effective and inexpensive herbicide in agriculture worldwide.[1⇓-3] However, PQ is highly toxic to humans and animals owing to its high toxicity,[4] which has aroused great concern from the public. PQ can be absorbed through a variety of ways, including oral, inhalational, and transdermal ways, leading to toxic effects on multiple organs.[5] PQ primarily accumulates in the lungs (6-10 times higher than plasma concentrations)[6] because it can be actively transported into lung alveolar epithelial cells and Clara cells by the polyamine uptake system.[7] Without effective antidotes, PQ-induced acute lung injury (ALI) and subsequent progressive pulmonary fibrosis are the primary causes of death.[8]
Although great attention has been given to PQ poisoning and some progress has also been made, the mechanisms of PQ toxicity remain to be further elucidated. Accumulated evidence has suggested that reactive oxygen species (ROS) play key roles in PQ poisoning. PQ, as an electron acceptor, can induce numerous ROS generated in cells,[9] which further leads to direct oxidative stress.[10] Additionally, ROS would also trigger cell apoptosis as signal transduction molecules.[11] Thus, direct ROS inhibition by antioxidants might be a potential effective way to ameliorate PQ injury. Mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs) and p38 MAPKs, are a group of serine-threonine protein kinases. MAPKs are vital in signal transduction from the cell surface to nucleus in response to a diverse array of extracellular stimuli.[12] In addition, MAPKs have been reported to be activated by ROS.[13] Moreover, it has been proven that suppressing the ROS/p38 MAPK pathway could reduce PQ-induced lung injury.[9,14]
Arctigenin, a natural lignan compound extracted from Arctium lappa L (A. Lappa), has attracted researchers’ attention for its promising therapeutic effects in the past several decades.[15] Numerous studies have reported the beneficial biological activities of arctigenin, including anti-oxidase,[16] anti-inflammation,[17] and antitumor activities.[17] More interestingly, our previous study demonstrated that arctigenin attenuated PQ-induced pulmonary fibrosis by suppressing epithelial-mesenchymal transition via the Wnt3a/β-catenin pathway.[18] Additionally, arctigenin has been shown to protect against lipopolysaccharide-induced lung injury in rats[19] and mice.[20] However, whether arctigenin can attenuate PQ-induced ALI and whether the ROS/p38 MAPK pathway is involved are still unknown. Thus, our present study was designed to investigate the effects of arctigenin on PQ-induced ALI and the underlying mechanisms.
METHODS
Cell culture
Human lung epithelial A549 cells were purchased from Fudan IBS Cell Center, Shanghai, China. Cells were cultured at 37 °C in 95% air and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) (HyClone, South Logan, USA) with 10% (v/v) fetal bovine serum (Invitrogen, Germany) and 1% (v/v) penicillin/streptomycin (Invitrogen, Germany). Subculture was performed with 0.25% trypsin/ethylenediaminetetraacetic acid (EDTA) solution (Invitrogen, Germany) after washing the cell monolayers twice with phosphate-buffered saline (PBS). Trypsin was then inactivated, and the cells were seeded into fresh T25 flasks.[21]
Cell viability assay
Cell viability was assayed using a cell counting kit-8 (CCK-8) kit (Beyotime, China) according to the manufacturer’s instructions.[22] Briefly, A549 cells were cultured in a 96-well plate and treated with PQ (Sigma-Aldrich, USA) at the indicated concentrations for 24 h with or without arctigenin (MedChemExpress, USA), while PBS was used as a control. After treatment, 10 μL of CCK-8 solution per well was added. The cells were then incubated at 37 °C for another 30 min, and the amount of formazan dye generated by cellular dehydrogenase activity was measured by absorbance at 450 nm with a microplate reader.
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labelling (TUNEL) assay
TUNEL assays were performed with the one-step TUNEL apoptosis assay kit (Beyotime, China) following the manufacturer’s instructions. Briefly, A549 cells were fixed in 4% paraformaldehyde in PBS and then permeabilized with 0.1% Triton X-100, followed by incubation with TUNEL working solution. The 4’,6-diamidino-2-phenylindole (DAPI) was used to stain and visualize all nuclei. Cy3-labelled TUNEL-positive cells were detected by fluorescence microscopy.
Mitochondrial membrane potential assay
Mitochondrial depolarization in A549 cells was measured using a 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethyl benzimidazolo-carbocyanine iodide (JC-1) probe (Beyotime, China) according to the information described previously.[23] In brief, cells were incubated with JC-1 staining solution (5 μg/mL) at 37 °C for 20 min, followed by rinsing with JC-1 staining buffer twice. JC-1, a cationic carbocyanine dye, exhibits potential-dependent accumulation in healthy mitochondria, where it forms J aggregates (red), while it remains a monomer (green) upon mitochondrial depolarization. The fluorescence of red aggregates and green monomers was captured by fluorescence microscopy, while the fluorescence intensity was quantified using ImageJ software.
ROS measurement
To detect the level of superoxide anions, the dihydroethidium (DHE) (Beyotime, China) staining was performed following the manufacturer’s instructions. A549 cells were incubated with 5 μmol/L DHE at 37 °C for 30 min and then rinsed in PBS three times. Thereafter, the cells were analyzed by fluorescence microscopy, and the red fluorescence intensity was quantified using ImageJ software. Additionally, the 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA) (Beyotime, China) assay was also carried out for the detection of oxidative species.
Immunoblotting studies
Immunoblotting studies were performed following procedures described in our previous reports with modifications.[21,24] In brief, proteins were extracted from A549 cells using RIPA buffer (Beyotime, China) containing protease inhibitor cocktail (Roche, USA) and phosphatase inhibitor cocktail (PhosSTOP™, Sigma-Aldrich, USA). Protein samples (50 μg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then electrotransferred to nitrocellulose membranes. Then, the blocked membranes were incubated at 4 °C overnight with primary antibodies, including rabbit antibodies against p38 MAPK, phospho-p38 MAPK, ERK, phospho-ERK, JNK, and phospho-JNK (1:1000, Cell Signaling Technology, USA). Rabbit anti-GAPDH or anti-β-actin antibody (1:3000, Abways, China) was used as an internal control. The membranes were washed and then incubated with horseradish peroxidase (HRP)-labelled goat anti-rabbit or goat anti-mouse secondary antibody (1:10000, Jackson ImmunoResearch, USA) at room temperature for 1 h. The membranes were washed three times in tris-buffered saline with Tween-20 (TBST), and the bands were detected using enhanced chemiluminescence reagents (Millipore, USA) with a Tanon 5200 Chemiluminescent Imaging System (Tanon Science & Technology, China). The images were analyzed by ImageJ software to obtain integrated intensities.
Statistical analysis
Data were presented as the mean±standard deviation (SD). Statistical significance was determined by analysis of variance (ANOVA) followed by a post hoc Tukey’s test for multigroup comparison. Differences were considered statistically significant when a P-value <0.05.
RESULTS
Arctigenin ameliorated PQ-induced A549 cell injury
To evaluate the protective effects of arctigenin in vitro, a PQ-induced A549 cell injury model was successfully established, and the results of the CCK-8 assay showed that A549 cell viability was inhibited by PQ in a dose-dependent manner (Figure 1A). Moreover, arctigenin showed no significant effect on the viability of A549 cells under control conditions (Figure 1B) but ameliorated PQ-induced inhibition of A549 cell viability in a dose-dependent manner (Figure 1C). Additionally, light microscopy indicated the protective effects of arctigenin on PQ-treated A549 cells, as reflected by cell number and morphology (Figure 1D). These data supported that arctigenin ameliorated PQ-induced A549 cell injury.
Figure 1.
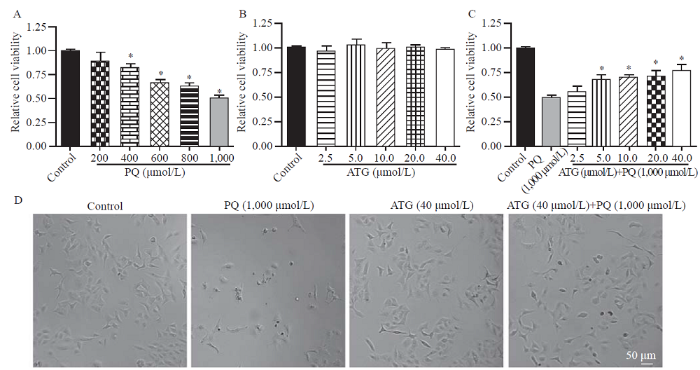
Figure 1.
Arctigenin ameliorated paraquat (PQ)-induced A549 cell injury. A: PQ inhibited the viability of A549 cells in a dose-dependent manner, reflected by cell counting kit-8 (CCK-8) assay (n=3, *P<0.05 vs. control); B: no dramatic effect of arctigenin was observed on the cell viability of A549 cells under control conditions, indicated by CCK-8 assay (n=3); C: the bar graph indicated arctigenin ameliorated PQ-induced A549 cell injury in a dose-dependent manner, determined by CCK-8 assay (n=3, *P<0.05 vs. PQ); D: representative light microscopy images indicated arctigenin ameliorated PQ-induced A549 cell injury, as reflected by more shrunken cells (scale bar=50 μm).
Arctigenin alleviated PQ-induced cell apoptosis
Cell death usually occurs upon toxic insults, and cell apoptosis plays an important role in PQ-induced lung injury.[25] Thus, we further tested the effects of arctigenin on PQ-induced A549 cell apoptosis by TUNEL assay and JC-1 staining. The results of the TUNEL assay showed that arctigenin dramatically reduced the number of TUNEL-positive cells induced by PQ (Figure 2A). Consistently, the JC-1 staining results also revealed that PQ-induced mitochondrial membrane potential loss, a marker for early-stage apoptosis, was significantly ameliorated by arctigenin treatment (Figure 2B). The above data demonstrated that arctigenin alleviated PQ-induced cell apoptosis.
Figure 2.
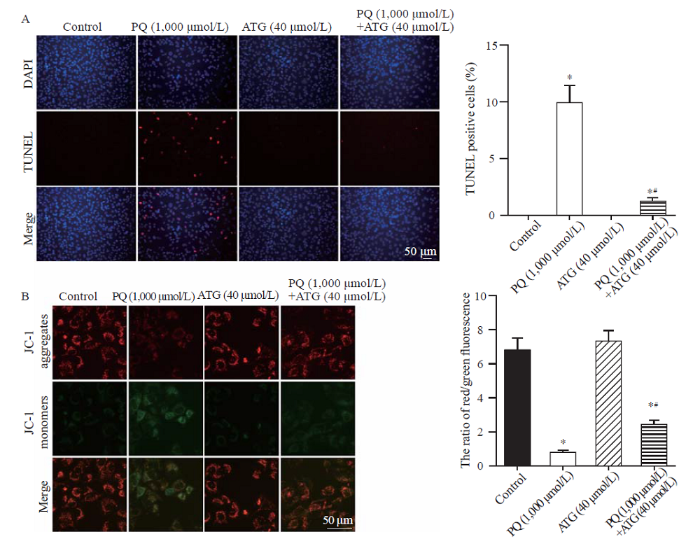
Figure 2.
Arctigenin alleviated paraquat (PQ)-induced A549 cell apoptosis. A: representative images and statistical results of TUNEL staining showing arctigenin reduced A549 cell apoptosis induced by PQ (n=5, *P<0.05 vs. control; #P<0.05 vs. PQ; scale bar=50 μm). B: representative pictures and statistical results indicating arctigenin alleviated PQ-induced mitochondrial membrane potential loss in A549 cells. JC-1 exhibited potential-dependent accumulation in healthy mitochondria where it formed J aggregates (red) while it remained as monomer (green) upon mitochondrial depolarization. The ratio of red/green fluorescence of JC-1 dramatically decreased in PQ-treated A549 cells, which was relieved by arctigenin co-administration (n=5, *P<0.05 vs. control; #P<0.05 vs. PQ; scale bar=50 μm).
Arctigenin reduced ROS generation in PQ-treated A549 cells
ROS are vital molecules in pathophysiological processes after PQ injury.[26,27] On the one hand, ROS can directly cause oxidative stress damage,[28] and on the other hand, ROS would serve as signaling molecules to trigger apoptosis.[29] Therefore, we explored the role of arctigenin on ROS generation in PQ-treated A549 cells by DHE staining and DCFH-DA staining. The representative images and statistical results indicated that the ROS level was dramatically increased in A549 cells after incubation with PQ for 24 h, but co-incubation with arctigenin remarkably inhibited PQ-induced ROS generation (supplementary Figures 1 A and B). These results showed that arctigenin reduced PQ-induced ROS generation in A549 cells.
Arctigenin inhibited p38 MAPK phosphorylation in PQ-treated A549 cells
Generally, increased ROS production in cells would lead to the activation of MAPKs, a family of serine/threonine protein kinases that mediate fundamental biological processes and cellular responses to external stress signals.[13,30] Moreover, the ROS/p38 MAPK pathway has been reported to participate in PQ-induced cell injury.[9,14] Accordingly, we further tested whether arctigenin would affect the activation of MAPKs, and the immunoblotting results showed that arctigenin significantly suppressed PQ-induced p38 MAPK phosphorylation (supplemental Figure 2A). In addition, PQ also increased JNK but inhibited ERK phosphorylation, on which arctigenin had little effect (supplementary Figures 2B and C). These data suggested that the inhibition of p38 MAPK was mainly involved in the protective effects of arctigenin.
DISCUSSION
Our study provides evidence that arctigenin protects against PQ-induced lung epithelial injury in vitro. Arctigenin ameliorated cell viability inhibition and cell apoptosis induced by PQ. Mechanistically, arctigenin may exert its protective effects by suppressing PQ-induced activation of ROS/p38 MAPK signaling (supplementary Figure 3).
Arctigenin is the main active ingredient from A. Lappa, a herbal medicine widely used in China. In recent years, arctigenin has been extensively studied for its pharmacological properties, and numerous studies have reported the beneficial effects of arctigenin on anti-inflammation,[31] antioxidative stress,[16,17] antitumor,[32] and antineuronal degeneration.[33] PQ poisoning has been frequently reported in recent years. As a highly toxic herbicide, PQ causes toxicity to multiple organs, including the lung, kidney, liver, and brain, among which the lung is the most targeted.[34] However, there is still little effective treatment for PQ-induced lung injury and subsequent fibrosis, which leads to high morbidity and frequently to death. A previous experiment has proven that arctigenin can ameliorate PQ-induced pulmonary fibrosis via suppressing the epithelial-mesenchymal transition.[18] Additionally, arctigenin has also been demonstrated to alleviate the lipopolysaccharide-induced lung injury in rats[19] and mice.[20] In line with these previous studies, our present work also supports the benefits of arctigenin against PQ-induced lung epithelial injury, as reflected by the CCK-8 cell viability assay. This finding implies that arctigenin also protects against PQ-induced ALI and the early stage of PQ-induced lung toxicity. Thus, arctigenin might be considered a potential candidate drug for the treatment of PQ-induced lung poisoning.
We next investigated the potential mechanisms by which arctigenin exerted its protective effect against PQ-induced lung epithelial injury. It is generally considered that cell death would occur upon toxic insults, and apoptosis is one of the classical ways by which PQ causes lung epithelial cell death.[24] Although arctigenin has been shown to induce apoptosis in several types of tumors and therefore has potential antitumor activity,[35] the effect of arctigenin on PQ-induced lung epithelial cell apoptosis remains unknown. Thus, we tested the role of arctigenin in PQ-induced A549 cell apoptosis via a TUNEL assay and mitochondrial membrane potential assay, and the results showed that arctigenin dramatically reduced PQ-induced A549 cell apoptosis, which is not consistent with what has been found previously in tumors. This may result from the fact that these are two categories of diseases, the tumors and the organ injuries, because other studies in myocardial ischemia-reperfusion injury[36] and brain injury[37] have also demonstrated the anti-apoptosis properties of arctigenin. However, the underlying mechanisms of these differences remain to be further explored.
ROS, including the superoxide anion (O2-), hydrogen peroxide (H2O2) and the hydroxyl radical (HO•), are a group of chemical species that are formed upon incomplete reduction of oxygen.[38] While PQ exerts its toxicity mainly through a process of redox cycling with superoxide anion production,[39] ROS generation and oxidative stress have a central role in PQ poisoning. More importantly, excessive ROS would trigger the apoptotic signaling pathway.[11,29] MAPKs, a group of serine-threonine protein kinases, play important roles in signal transduction in response to diverse extracellular stimuli.[12] MAPKs have also been reported to be activated by ROS.[13] In addition, it has been proven that PQ-induced activation of p38 MAPK plays a key role in the apoptotic pathway of A549 cells and that inhibition of ROS/p38 signaling could ameliorate PQ-induced lung injury.[9,14] Thus, in our present study, we further checked whether ROS/p38 MAPK signaling was involved in the protective effect of arctigenin against PQ-induced lung epithelial injury. We found that arctigenin remarkably reduced PQ-induced ROS generation and p38 MAPK phosphorylation. This finding supports the previous concept that ROS/p38 signaling is vital in PQ toxicity.
However, we also realized that one of the limitations of our present work was the lack of in vivo experiments since only human lung epithelial A549 cells were used. It should be noted that what has been found in these cell experiments might not be equally applicable in animals or even in the clinic; thus, it would be more stringent to use in vivo studies to verify the protective effects of arctigenin.
CONCLUSIONS
Our present study shows that arctigenin attenuates PQ-induced lung epithelial A549 cell injury in vitro by suppressing PQ-activated ROS/p38 MAPK-mediated cell apoptosis, and arctigenin might be considered a potential candidate drug for PQ-induced ALI.
Funding: This work was supported by the National Natural Science Foundation of China (82172182 and 82102311); Social Development Projects of Jiangsu Province (BE2017720); Natural Science Foundation of Jiangsu Province (BK20190247); Science Foundation of Jiangsu Health Commission (H2018039); and Jiangsu Postdoctoral Research Foundation (2018K048A and 2020Z193).
Ethical approval: No animal or human subjects were involved.
Conflicts of interest: None.
Contributors: CL, ZRS, and MMW performed the experiments; ZZY, WZ, and YR provided help during the experiments; XQH, RL, and QL contributed to the manuscript revision and data curation; SNN supervised all experiments.
All the supplementary files in this paper are available at http://wjem.com.cn.
Reference
Molecular mechanisms of paraquat-induced acute lung injury: a current review
DOI:10.3109/01480545.2013.834361 URL [Cited within: 1]
Evaluation of gastric lavage efficiency and utility using a rapid quantitative method in a swine paraquat poisoning model
DOI:10.5847/wjem.j.1920-8642.2020.03.008 URL [Cited within: 1]
Role of epithelial-to-mesenchymal transition in the pulmonary fibrosis induced by paraquat in rats
DOI:10.5847/wjem.j.1920-8642.2021.03.009 URL [Cited within: 1]
Role of antioxidants in paraquat toxicity
DOI:10.1016/S0300-483X(02)00382-7 URL [Cited within: 1]
Paraquat poisoning mechanism and its clinical treatment progress
Paraquat poisonings: mechanisms of lung toxicity, clinical features, and treatment
PMID:18161502
[Cited within: 1]

Paraquat dichloride (methyl viologen; PQ) is an effective and widely used herbicide that has a proven safety record when appropriately applied to eliminate weeds. However, over the last decades, there have been numerous fatalities, mainly caused by accidental or voluntary ingestion. PQ poisoning is an extremely frustrating condition to manage clinically, due to the elevated morbidity and mortality observed so far and due to the lack of effective treatments to be used in humans. PQ mainly accumulates in the lung (pulmonary concentrations can be 6 to 10 times higher than those in the plasma), where it is retained even when blood levels start to decrease. The pulmonary effects can be explained by the participation of the polyamine transport system abundantly expressed in the membrane of alveolar cells type I, II, and Clara cells. Further downstream at the toxicodynamic level, the main molecular mechanism of PQ toxicity is based on redox cycling and intracellular oxidative stress generation. With this review we aimed to collect and describe the most pertinent and significant findings published in established scientific publications since the discovery of PQ, focusing on the most recent developments related to PQ lung toxicity and their relevance to the treatment of human poisonings. Considerable space is also dedicated to techniques for prognosis prediction, since these could allow development of rigorous clinical protocols that may produce comparable data for the evaluation of proposed therapies.
Putrescine and 5-hydroxytryptamine accumulation in rat lung slices: cellular localization and responses to cell-specific lung injury
DOI:10.1016/0041-008X(87)90198-0 URL [Cited within: 1]
Aspirin-triggered resolvin D1 alleviates paraquat-induced acute lung injury in mice
DOI:10.1016/j.lfs.2018.12.028 URL [Cited within: 1]
Aspirin eugenol ester ameliorates paraquat-induced oxidative damage through ROS/p38-MAPK-mediated mitochondrial apoptosis pathway
DOI:10.1016/j.tox.2021.152721 URL [Cited within: 4]
Role of oxidative stress in nanoparticles toxicity
DOI:10.1080/10715762.2020.1859108 URL [Cited within: 1]
Role of reactive oxygen species (ROS) in apoptosis induction
DOI:10.1023/a:1009616228304
PMID:11256882
[Cited within: 2]
Reactive oxygen species (ROS) and mitochondria play an important role in apoptosis induction under both physiologic and pathologic conditions. Interestingly, mitochondria are both source and target of ROS. Cytochrome c release from mitochondria, that triggers caspase activation, appears to be largely mediated by direct or indirect ROS action. On the other hand, ROS have also anti-apoptotic effects. This review focuses on the role of ROS in the regulation of apoptosis, especially in inflammatory cells.
Mitogen-activated protein kinases in normal and (pre)neoplastic ovarian surface epithelium
DOI:10.1186/1477-7827-1-71 URL [Cited within: 2]
Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways
Klotho alleviates lung injury caused by paraquat via suppressing ROS/P38 MAPK-Regulated inflammatory responses and apoptosis
Overview of the anti-inflammatory effects, pharmacokinetic properties and clinical efficacies of arctigenin and arctiin from Arctium lappa L
DOI:10.1038/aps.2018.32 URL [Cited within: 1]
Arctigenin attenuates ischemia/reperfusion induced ventricular arrhythmias by decreasing oxidative stress in rats
DOI:10.1159/000493038 URL [Cited within: 2]
Molecular mechanisms of the action of Arctigenin in cancer
DOI:10.1016/j.biopha.2018.08.158 URL [Cited within: 3]
Arctigenin suppressed epithelial-mesenchymal transition through Wnt3a/β-catenin pathway in PQ-induced pulmonary fibrosis
DOI:10.3389/fphar.2020.584098 URL [Cited within: 2]
Arctigenin attenuates lipopolysaccharide-induced acute lung injury in rats
DOI:10.1007/s10753-014-9969-z URL [Cited within: 2]
Arctigenin protects against lipopolysaccharide-induced pulmonary oxidative stress and inflammation in a mouse model via suppression of MAPK, HO-1, and iNOS signaling
DOI:10.1007/s10753-015-0115-3 URL [Cited within: 2]
MG53 protects against contrast-induced acute kidney injury by reducing cell membrane damage and apoptosis
DOI:10.1038/s41401-020-0420-8 URL [Cited within: 2]
Gastrin attenuates renal ischemia/reperfusion injury by a PI3K/Akt/bad-mediated anti-apoptosis signaling
DOI:10.3389/fphar.2020.540479 URL [Cited within: 1]
Simvastatin mitigates apoptosis and transforming growth factor-beta upregulation in stretch-induced endothelial cells
Increased AT1 receptor expression mediates vasoconstriction leading to hypertension in Snx1-/- mice
DOI:10.1038/s41440-021-00661-x URL [Cited within: 2]
Paraquat induces lung alveolar epithelial cell apoptosis via Nrf-2-regulated mitochondrial dysfunction and ER stress
DOI:10.1007/s00204-012-0873-8
PMID:22678742
[Cited within: 1]
Paraquat (1,1'-dimethyl-4,4'-bipyridinium chloride; PQ) is widely and commonly used as a herbicides in the world. PQ has been reported to be a major hazard because it causes lung injury. However, the molecular mechanisms underlying PQ-induced lung toxicity still need to be elucidated. Here, we found that PQ significantly decreases cell viability, increases sub-G1 hypodiploids DNA contents and caspase 3/7 activity in lung alveolar epithelial cell-derived L2 cells, which also caused mitochondrial dysfunction, and decreased the mRNA expression of Bcl-2 and increased that of Bax, Bak, and p53. Moreover, the protein expressions of Bax and Bak were increased in PQ-treated cells. In addition, when PQ was exposed to L2 cells, the expressions of ER stress-related signaling genes (including Grp78, CHOP, and caspase-12 mRNA) and proteins (including phospho-eIF-2α, CHOP, Grp78, calpain I and -II, and caspase-12) were significantly increased. PQ also decreased the protein expressions of pro-caspase-9/7/3. Next, we investigated the role of Nrf-2 in PQ-induced alveolar epithelial cell toxicity. In L2 cells, PQ induced Nrf-2 translocation from the cytosol to the nucleus. Cells transfected with Nrf-2 siRNA significantly reversed the PQ-induced toxicity, including depolarization of MMP, increased the Bax, Bak, p53 mRNAs expression, decreased the Bcl-2 mRNA expression, increased the caspase 3/7 activity, Grp78, CHOP, and caspase-12 mRNAs and protein expression, and decreased that of pro-caspase-3. Taken together, these results suggest that Nrf-2-regulated mitochondria and ER stress-related pathways are involved in the PQ-induced alveolar epithelial cell injury.
Paraquat induced DNA damage by reactive oxygen species
DOI:10.1080/15216549600201061 URL [Cited within: 1]
Mechanism of cytotoxicity of paraquat
DOI:10.1265/ehpm.2002.89 URL [Cited within: 1]
Reactive oxygen species: oxidative damage and pathogenesis
Signalling apoptosis: a radical approach
DOI:10.1179/135100001101536085
PMID:11450987
[Cited within: 2]
Reactive oxygen species (ROS) are frequently associated with cytotoxicity, often being described as damaging, harmful or toxic. It is generally assumed that, under pathological circumstances, ROS elicit wide-spread and random acts of oxidation. This passive attack of cellular components by ROS, in conditions where oxidative stress is the initiating stimulus for apoptosis, is assumed to simply trigger cell death as a result of cumulative oxidative damage. However, accumulating evidence now suggests that ROS may act as signalling molecules for the initiation and execution of the apoptotic death programme in many, if not all, current models of apoptotic cell death. Signalling by ROS would not appear to be random, as previously assumed, but targeted at specific metabolic and signal transduction cellular components. There is also evidence that the enzymatic generation of ROS may not simply be an unwanted by-product of the primary reaction catalysed, but that ROS may be used as signalling molecules to regulate cellular processes including apoptosis. This view of ROS as signalling molecules (as opposed to toxic metabolites) has been further bolstered by the findings that cellular antioxidants such as glutathione and thioredoxin not only serve to regulate ROS levels but also act as reversible redox modifiers of enzyme function. This review will attempt to delineate the involvement of ROS in apoptosis in light of these recent discoveries and provide evidence for a crucial role for ROS in the initiation and execution of the death process.
MAPK signalling pathways as molecular targets for anti-inflammatory therapy—from molecular mechanisms to therapeutic benefits
PMID:16198162
[Cited within: 1]
Excessive inflammation is becoming accepted as a critical factor in many human diseases, including inflammatory and autoimmune disorders, neurodegenerative conditions, infection, cardiovascular diseases, and cancer. Cerebral ischemia and neurodegenerative diseases are accompanied by a marked inflammatory reaction that is initiated by expression of cytokines, adhesion molecules, and other inflammatory mediators, including prostanoids and nitric oxide. This review discusses recent advances regarding the detrimental effects of inflammation, the regulation of inflammatory signalling pathways in various diseases, and the potential molecular targets for anti-inflammatory therapy. Mitogen-activated protein kinases (MAPKs) are a family of serine/threonine protein kinases that mediate fundamental biological processes and cellular responses to external stress signals. Increased activity of MAPK, in particular p38 MAPK, and their involvement in the regulation of the synthesis of inflammation mediators at the level of transcription and translation, make them potential targets for anti-inflammatory therapeutics. Inhibitors targeting p38 MAPK and JNK pathways have been developed, and preclinical data suggest that they exhibit anti-inflammatory activity. This review discusses how these novel drugs modulate the activity of the p38 MAPK and JNK signalling cascades, and exhibit anti-inflammatory effects in preclinical disease models, primarily through the inhibition of the expression of inflammatory mediators. Use of MAPK inhibitors emerges as an attractive strategy because they are capable of reducing both the synthesis of pro-inflammatory cytokines and their signalling. Moreover, many of these drugs are small molecules that can be administered orally, and initial results of clinical trials have shown clinical benefits in patients with chronic inflammatory disease.
Arctigenin ameliorates inflammation in vitro and in vivo by inhibiting the PI3K/AKT pathway and polarizing M1 macrophages to M2-like macrophages
DOI:10.1016/j.ejphar.2013.01.014 URL [Cited within: 1]
Arctigenin attenuates tumor metastasis through inhibiting epithelial-mesenchymal transition in hepatocellular carcinoma via suppressing GSK3β-dependent Wnt/β-catenin signaling pathway in vivo and in vitro
DOI:10.3389/fphar.2019.00937 URL [Cited within: 1]
Arctigenin effectively ameliorates memory impairment in Alzheimer’s disease model mice targeting both β-amyloid production and clearance
DOI:10.1523/JNEUROSCI.4790-12.2013 URL [Cited within: 1]
Evidence for energy-dependent accumulation of paraquat into rat lung
Arctigenin induces the apoptosis of primary effusion lymphoma cells under conditions of glucose deprivation
AMPK/SIRT1 pathway is involved in arctigenin-mediated protective effects against myocardial ischemia-reperfusion injury
DOI:10.3389/fphar.2020.616813 URL [Cited within: 1]
Arctigenin treatment protects against brain damage through an anti-inflammatory and anti-apoptotic mechanism after needle insertion
DOI:10.3389/fphar.2016.00182
PMID:27445818
[Cited within: 1]
Convection enhanced delivery (CED) infuses drugs directly into brain tissue. Needle insertion is required and results in a stab wound injury (SWI). Subsequent secondary injury involves the release of inflammatory and apoptotic cytokines, which have dramatic consequences on the integrity of damaged tissue, leading to the evolution of a pericontusional-damaged area minutes to days after in the initial injury. The present study investigated the capacity for arctigenin (ARC) to prevent secondary brain injury and the determination of the underlying mechanism of action in a mouse model of SWI that mimics the process of CED. After CED, mice received a gavage of ARC from 30 min to 14 days. Neurological severity scores (NSS) and wound closure degree were assessed after the injury. Histological analysis and immunocytochemistry were used to evaluated the extent of brain damage and neuroinflammation. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was used to detect universal apoptosis. Enzyme-linked immunosorbent assays (ELISA) was used to test the inflammatory cytokines (tumor necrosis factor (TNF)-alpha, interleukin (IL)-6 and IL-10) and lactate dehydrogenase (LDH) content. Gene levels of inflammation (TNF-alpha, IL-6, and IL-10) and apoptosis (Caspase-3, Bax and Bcl-2) were detected by reverse transcription-polymerase chain reaction (RT-PCR). Using these, we analyzed ARC's efficacy and mechanism of action. Results: ARC treatment improved neurological function by reducing brain water content and hematoma and accelerating wound closure relative to untreated mice. ARC treatment reduced the levels of TNF-alpha and IL-6 and the number of allograft inflammatory factor (IBA)- and myeloperoxidase (MPO)-positive cells and increased the levels of IL-10. ARC-treated mice had fewer TUNEL+ apoptotic neurons and activated caspase-3-positive neurons surrounding the lesion than controls, indicating increased neuronal survival. Conclusions: ARC treatment confers neuroprotection of brain tissue through anti-inflammatory and anti-apoptotic effects in a mouse model of SWI. These results suggest a new strategy for promoting neuronal survival and function after CED to improve long-term patient outcome.
ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis
New insights into antioxidant strategies against paraquat toxicity
DOI:10.3109/10715762.2014.899694 URL [Cited within: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}