


INTRODUCTION
Acute pulmonary embolism (APE) is a syndrome that causes pulmonary circulation disturbance due to endogenous or exogenous embolus blocking the main stem or branch of pulmonary artery.[1,2] It is the third most common cardiovascular disease with an annual incidence rate from 39 to 115 per 100,000 population.[1] The mortality rate of cardiac arrest (CA) caused by APE is as high as 65%-95%.[3] The main rescue measures for CA patients with suspected APE are cardiopulmonary resuscitation (CPR) and intravenous thrombolysis.[1] The main cause of death is pulmonary arterial hypertension (PAH) following APE, which leads to acute right heart failure.[4] The pathological and physiological characteristics of PAH are abnormal pulmonary vascular contraction and vascular remodeling in chronic pulmonary thromboembolism.[5] Early inhibition of pulmonary vascular remodeling may reduce acute persistent PAH and hold up the formation of chronic PAH following APE. The pulmonary artery remodeling is initiated by pulmonary vascular endothelial cell dysfunction.[6] The pathological manifestations of this dysfunction include endothelial cell proliferation and concurrent neoangiogenesis, leading to the formation of glomeruloid structures known as plexiform lesions.[6] However, in massive APE, whether early pulmonary vascular remodeling has initiated and what are the pathological manifestations on pulmonary vascular endothelial cell remain to be established.
Endothelial dysfunction is caused by an imbalance in endothelial cell production of vasoconstrictors and vasodilators.[7] In the renin angiotensin system (RAS), angiotensin-converting enzyme (ACE)-angiotensin (Ang) II-Ang II type 1 receptor (AT1) axis comprises vasoconstrictors that promote cell growth and vascular remodeling in PAH.[8] The recently discovered ACE2-Ang (1-7)-Mas receptor axis of the RAS contributes to vasodilation and inhibits apoptosis, angiogenesis, and proliferation.[9] Imbalance of the ACE2 and ACE axes is an important factor in endothelial dysfunction and vascular pathology.[10] Whether the ACE and ACE2 axes participate in endothelial dysfunction during the early pulmonary arterial remodeling caused by massive APE with CA is not clear.
The ACE inhibitor captopril is a vasodilator that prevents the conversion of Ang I to Ang II. The result from our previous study confirmed that the pulmonary vascular resistance was reduced and the ACE2/ACE axes ratio was increased by captopril after ROSC in APE.[11] To test the hypothesis that increasing ACE2/ACE axes ratio alleviates the early pulmonary arterial remodeling in APE, we examined the endothelial pathological changes and expression of ACE2/ACE axes in pulmonary artery with or without captopril in a porcine massive APE with CA model.
METHODS
Ethical approval
The experimental procedures were permitted by the Capital Medical University Institutional Animal Care Committee (permit number: 2010-D-013). All animals were housed individually in cages (80 cm×80 cm×90 cm) and fed with standard pig chow and free access to water. We kept room temperature at 26 ℃ and body temperature at 37 ℃ using an infrared lamp.
Animal preparation
All procedures were performed on Landrace pigs, because of their similarity to humans in terms of the pathological and physiological characteristics of cardiovascular and endocrine diseases. Twenty-nine pigs from Beijing Experimental Animal Center (license number: SCXK 11-00-002) were used (both sexes, age 3 months, weight 28±2 kg). The midazolam (0.2 mg/kg) was used for intramuscular sedation, and then 3% pentobarbital (8 mg/[kg·h]) was intravenous continuously after propofol (1.0 mg/kg) injection through ear vein for general anesthesia. All pigs received mechanical ventilation by a ventilator (Evita 4, Draeger Medical, Germany) which delivered a tidal volume of 8 mL/kg at a respiratory rate of 12 to 20 breaths/min after endotracheal intubation. The end-tidal CO2 was maintained at 30-40 mmHg (1 mmHg=0.133 kPa) and oxygen saturation (SpO2) 95%-100%. Central venous pressure was monitored through a triple-lumen central venous catheter inserting into the left femoral vein. Aortic pressure was observed through an arterial catheter inserting into the left femoral artery. Mean pulmonary artery pressure was measured by a Swan-Ganz catheter (7-Fr, Edwards Life Sciences, USA) inserting into the pulmonary artery through the right external jugular vein. All hemodynamic parameters were recorded under an M8001A monitor (Philips Medizin Systeme Boeblingen GmbH, Germany). The central venous pressure was maintained at 5-12 mmHg by infusion of normal saline (8 mL/[kg·h]) through the right femoral vein. A large-bore catheter (1.0-cm internal diameter) was manipulated into the left external jugular vein under the guidance of computed tomography (CT) so that its tip was finally positioned very close to the orifice of the pulmonary artery trunk. The M8001A monitor (Philips Medizin Systeme Boeblingen GmbH, Germany) was also used to visualize the electrocardiogram. All wounds were partially sutured. Catheter tips were left outside the body after the operation.
Experimental protocol
After penetration of the femoral vein, 100 mL of blood was obtained and left at room temperature for 2-3 h for self-coagulation. About 10-15 mL blood clots were chopped into small clots (1.5 cm×1.0 cm×1.0 cm) which were then suspended in normal saline in a large catheter-tip syringe. The suspension was then injected into the left external jugular vein over the next 2 min, until mean arterial pressure (MAP) dropped below 30 mmHg.[12]Animals were injected with 15,000 U/kg urokinase (Nanjing Nanda Pharmaceutical Co., Ltd., China) through a Swan-Ganz catheter that had been inserted into the pulmonary artery and received CPR (as dictated by the 2010 American Heart Association guidelines). If systolic blood pressure remained >50 mmHg for more than 10 consecutive minutes, we considered the animals to obtain the ROSC.[13]Pigs were considered dead if spontaneous circulation was not restored within 30 min. CT-guided pulmonary angiography was used to diagnose pulmonary embolism.
Twenty-nine animals were randomly divided into the following four groups. In the control group (n=5), the 10 mL of potassium chloride (15%) was injected intravenously into pigs after a bolus of propofol (3.0 mg/kg, 6 h after experimental preparation). In the APE-CA group (n=5), pigs were injected quickly with thrombus until MAP dropped below 30 mmHg.[12] Nineteen animals received thrombus injections until MAP dropped below 30 mmHg and then were treated with urokinase and CPR. Ten successfully resuscitated animals were randomly divided into two groups. In the ROSC-captopril group (n=5), animals received captopril (Sigma-Aldrich, C8856-1G) with 22.22 mg/kg intravenously at 30 min after ROSC.[14] Two pigs died at 6 h after ROSC. In the ROSC-saline group (n=5), the animals were injected with the same volume of saline (SA) at 30 min after ROSC. One pig died at 4 h after ROSC, and another died 6 h after ROSC. At 6 h after ROSC, animals were euthanized with potassium chloride (15%, 10 mL) intravenously following injection of propofol with 3.0 mg/kg.
Histological evaluation
Pulmonary artery samples obtained at 6 h after ROSC were fixed with 10% buffered formalin, embedded in paraffin, sectioned with 5-μm thickness and processed for routine hematoxylin and eosin staining.
Western blotting analysis
Pulmonary artery tissue was homogenized in radio-immunoprecipitation assay (RIPA) lysis buffer plus protease inhibitors (Roche, Switzerland) and phosphatase inhibitors (Roche, Switzerland). About 20 mg of the tissue was cut into small pieces and homogenized in RIPA lysis buffer (200 µL). Tissue lysate was separated by centrifugation with 13,000×g for 20 min at 4 °C, and swiftly harvested and kept in reserve at -80 °C. The total protein concentration was measured with the bicinchoninic acid (BCA) assay (Pierce, USA). Homogenates were heated in the sample buffer at 95 °C for 5 min. The total protein (40 μg) was then run on the 10% and 12% sodium dodecyl sulfate (SDS)-polyacrylamide gels and wet-transferred to a polyvinylidene fluoride membrane (0.45 μm, Millipore, USA). Blots were blocked using 5% nonfat dry milk in tris-buffered saline with Tween-20 (TBST) buffer (1 mol/L Tris-HCl [pH 7.5], 100 mmol/L NaCl, 20% Tween 20) for 2 h and incubated overnight at 4 °C with 1:1000 B-cell lymphoma-2 (Bcl-2), Bcl-2-associated X (Bax), angiopoietin-2, Tie 2, or 1:5000 cleaved caspase-3 (Cell Signaling Technology, Inc., USA), or 1:10000 angiopoietin-1 antibody (Santa Cruz Biotechnology, Inc., USA), vascular endothelial growth factor (VEGF), β-actin antibody (Abcam, UK). Following incubation, the blots were washed in TBST, and then incubated for 40 min at room temperature with secondary anti-rabbit or anti-mouse IgG-horseradish peroxidase-linked antibody (1:10000, Jackson ImmunoResearch Laboratories, Inc., USA). Bands were detected using enhanced chemiluminescence (Millipore, USA). The optical density for quantification was obtained with Gel Image system ver.4.0 (Tanon Science & Technology Co., Ltd., China).
Immunohistochemical staining
The 3-μm-thickness pulmonary artery tissue embedded in paraffin was deparaffinized and serially dehydrated in ethanol. After blocking with 5% bovine serum albumin for 4 h, sections were then incubated overnight at 4 ℃ with anti-Bax (anti-mouse, 1: 250, Santa Cruz Biotechnology, Inc., USA), anti-angiopoietin-2 (anti-rabbit, 1:200, Abcam, UK), and anti-VEGF (anti-rabbit,1:200, Abcam, UK). The sections were washed with phosphate-buffered saline (PBS) (5 min, 3 times) and incubated with biotinylated secondary antibodies (1:200) for 2 h at room temperature, followed by incubation with avidin-biotin-peroxidase solution. The primary antibody with normal rabbit serum served as negative controls. Peroxidase conjugate localization was determined by diaminobenzidine tetrahydrochloride (DAB, Sigma, USA) as the chromogen and counterstained with hematoxylin. The positive areas showed the color of brown-yellow. Sections were viewed using an IX80 microscopy (Olympus, Japan) and analyzed using Image-Pro Insight 6.0 software (Media Cybernetics, USA).
Ultrastructure evaluation
Ultrastructure of pulmonary artery endothelial cells was observed by electron microscopy. Samples were obtained within 1-2 min of sacrifice. Pulmonary artery tissues were sliced (approximately 1-mm thickness) on ice, and then fixed for 30 min at 4 ℃ with glutaraldehyde (2.5%). Samples were postfixed for 1 h with 1% (v/v) osmic acid, then dehydrated and embedded using Epon812 at 40 °C for 4 h, 50 °C for 2 h, or 90 °C for 12 h. Sections (0.5-1.0 μm thickness) were cut and stained using toluidine blue (0.5%), and then observed using a light microscope (Nikon Corporation, Japan). Ultra-thin (approximately 60 nm) sections were double-stained using uranyl acetate (2.0%) for 20 min and lead citrate (2.0%) for 15 min, and then examined by blinded observers under a HITACHI HT7700 transmission electron microscope (Hitachi Scientific Instruments, USA).
Statistical analysis
The results were expressed as mean±standard deviation (SD). The difference between groups was calculated by Student's t-test or analysis of variance (ANOVA). Pearson correlation was used to analyze correlations between the ACE2/ACE axes ratio and angiopoietin, ACE2/ACE axes ratio and VEGF, ACE2/ACE axes ratio and Bcl-2/Bax, ACE2/ACE axes ratio and cleaved caspase-3. P<0.05 was considered statistically significant. All the analyses were conducted using the SPSS 25.0 software (SPSS Inc, USA).
RESULTS
Proliferative and apoptotic changes of endothelial cells in the APE-CA model
As shown in Figure 1A, the pulmonary arteries were almost completely occluded by accumulated endothelial cells in the APE-CA and ROSC-saline groups, while no angiostenosis due to endothelial cell proliferation was displayed in the control group. Fewer clogged arteries were discovered in the ROSC-captopril group than in the ROSC-saline group. The arteriolar lesions were characterized by plexiform lesion and concentric lesion composed of endothelial cells in “onion-skin” layers in APE (Figure 1A). Electron microscopy was used to observe the endothelial cells apoptosis and migration into vascular lumen in the APE-CA group, unlike the normal endothelial cell structure in the control group (Figure 1B).
Figure 1.

Figure 1.
Morphological and ultrastructure evaluation of pulmonary artery in APE. A: morphological evaluation of pulmonary artery in APE. Pulmonary arteries were occluded in the APE-CA and ROSC-saline groups (black arrow, ×100); fewer pulmonary arteries were occluded in the ROSC-captopril group (×100); plexiform lesions in the ROSC-saline group (enlargement, black arrows, ×400) and concentric lesions in ROSC-captopril group (enlargement, black arrows, ×400) were found. B: ultrastructure of endothelial cells in APE. Normal endothelial cell structure in the control group (black arrows, ×7,000), endothelial cell migration into vascular lumen in the APE-CA group (black arrows, ×10,000), and endothelial cell apoptosis in the APE-CA group (black arrows, ×10,000 and ×30,000). APE: acute pulmonary embolism; ROSC: return of spontaneous circulation; CA: cardiac arrest.
Pulmonary arterial expression of ACE2/ACE axes ratio after APE-CA
Compared with the control group, at the time of CA caused by APE, expression levels of ACE (P=0.001) and AT1 receptor (P<0.001) in the pulmonary artery increased, while the expression of ACE2 and Mas receptor declined dramatically (P=0.019, 0.029, respectively; Figure 2A). The expression of ACE (P=0.004) and ACE2 (P=0.001) declined significantly in the ROSC-saline group, compared to the APE-CA group (Figure 2A). To confirm the opposing effects of the ACE2-Ang (1-7)-Mas receptor axis and the ACE-Ang II-AT1 receptor axis in massive APE, the correlation between the two axes was analyzed in the control group, APE-CA group, and ROSC-saline group. The results showed positive correlations between ACE2 and Mas receptor (r=0.748, P=0.005), and between ACE and AT1 receptor (r=0.716, P=0.009). Negative correlations were observed between Mas and AT1 receptors (r= -0.665, P=0.018). Immunohistochemical staining showed that ACE2 and Mas receptor were expressed in vascular endothelial cells and abnormally proliferative endothelial cells, which formed plexiform lesions (Figure 2B).
Figure 2.
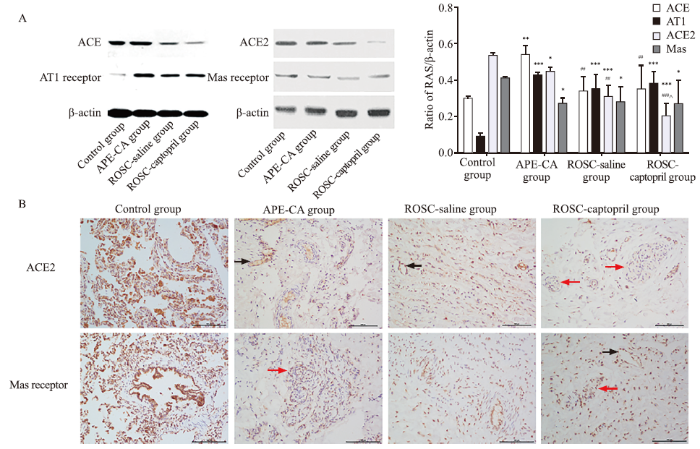
Figure 2.
Protein expression of RAS in the pulmonary artery and effects of captopril on APE. A: protein expression and quantitative analysis of RAS in the pulmonary artery and effects of captopril on APE, as revealed by Western blotting analysis; B: immunohistochemistry analysis of protein expression of ACE2 in the pulmonary artery in the control, APE-CA (black arrow pointing to expression of ACE2 in vascular endothelial cells), ROSC-saline (black arrow showing expression of ACE2 in vascular endothelial cells), and ROSC-captopril groups (red arrow showing expression of ACE2 in proliferative endotheliocytes); protein expression of Mas receptor in the pulmonary artery in the control, APE-CA (red arrow showing expression of Mas in proliferative endotheliocytes), ROSC-saline, and ROSC-captopril groups (black arrow showing expression of Mas receptor in vascular endothelial cells; red arrow showing expression of Mas receptor in proliferative endotheliocytes). Compared with the control group, *P<0.05, **P<0.01, ***P<0.001; compared with the APE-CA group, ##P<0.01, P<0.001; compared with the ROSC-saline group, ^P<0.05. Scale bar in B: 100 μm. RAS: renin angiotensin system; APE: acute pulmonary embolism; ACE2: angiotensin-converting enzyme 2; CA: cardiac arrest; ROSC: return of spontaneous circulation.
Pulmonary artery expression of angiopoietin-2/angiopoietin-1, VEGF, and Bax in APE-CA
The angiopoietin-2/angiopoietin-1 ratio increased 2.80 fold in the APE-CA group compared with that in the control group (P<0.05; supplementary Figure 1A). There was no significant difference in p-Tie 2 expression between groups. The expression of VEGF and Bax was significantly higher in the APE-CA and ROSC-saline groups than in the control group (all P<0.01; supplementary Figures 1 A and B). Compared with the control group, cleaved caspase-3 levels were higher in the ROSC-saline groups (P<0.001; supplementary Figure 1B). Correlation analysis showed that levels of angiopoietin-1 and angiopoietin-2 presented opposing trends (r= -0.747, P=0.008), while angiopoietin-2 and VEGF expression were coincident (r=0.927, P<0.001). Immunohistochemical staining showed that Bax was expressed in pulmonary artery endothelial cells (supplementary Figure 2A); angiopoietin-2 (supplementary Figure 2B) and VEGF (supplementary Figure 2C) were expressed in the abnormally proliferative endothelial cells of plexiform lesions during APE.
Effect of increasing ACE2/ACE axes ratio on post-resuscitation angiopoietin-2/angiopoietin-1 and VEGF expression in APE-CA
In contrast to the APE-CA group, the ROSC-saline group showed a slight decrease in angiopoietin-2/angiopoietin-1 ratio (P>0.05) and a substantial decrease after captopril treatment (P<0.05; supplementary Figure 1A). Similarly, captopril inhibited VEGF activation compared with saline after ROSC (P<0.05; supplementary Figure 1A). Correlations between ACE2/ACE axes ratio and angiopoietins/VEGF were analyzed. There were positive correlations between ACE and angiopoietin-2 (r=0.645, P=0.032), and between ACE and VEGF (r=0.612, P=0.045). There were negative correlations between ACE and angiopoietin-1 (r= -0.665, P=0.026), ACE2/ACE axes ratio and angiopoietin-2 (r= -0.833, P=0.001), and between ACE2/ACE axes ratio and VEGF (r= -0.893, P<0.001).
Effect of increasing ACE2/ACE axes ratio on post-resuscitation apoptosis of endothelial cells in APE
In the APE-CA group, there were significant correlations between Bcl-2/Bax and ACE2/ACE axes ratio (r=0.981, P<0.001), and between cleaved caspase-3 and ACE2/ACE axes ratio (r= -0.602, P=0.038). Captopril increased the Bcl-2/Bax ratio (P=0.006) and decreased cleaved caspase-3 levels (P=0.004; supplementary Figure 1B).
DISCUSSION
In this study, we described endothelial proliferation and apoptosis during early pulmonary arterial remodeling and imbalance of the ACE2 and ACE axes and increases in the angiopoietin-2/angiopoietin-1 ratio, VEGF, and pro-apoptotic factors. Increasing ACE2/ACE axes ratio with captopril could inhibit the activation of angiopoietin-2/angiopoietin-1, VEGF, and pro-apoptotic factors after ROSC. This finding implicates a critical role for increasing ACE2/ACE axes ratio in alleviating endothelial proliferation and apoptosis during the early pulmonary arterial remodeling of acute PAH due to massive APE.
The endothelial dysfunction triggers vascular remodeling, which leads to the formation of PAH.[15] The pathological characteristics of pulmonary endothelial dysfunction in PAH include disordered proliferation, abnormal intimal thickening, and the formation of fibrotic, plexiform, concentric, and dilation/angiomatoid lesions. Plexiform lesions are formed by irregular endothelial cell masses and sparse myofibroblasts. Concentric lesions are composed of endothelial cells and myofibroblasts in “onion-skin” layers.[16] These findings are in partial accordance with our morphological analysis of the pulmonary artery in the APE-CA model which showed that endothelial cells proliferated randomly, forming plexiform and concentric lesions. In addition, electron microscopy revealed endothelial cell apoptosis. These results illustrate that endothelial cell apoptosis and proliferation contribute to early pulmonary arterial remodeling in APE.
Besides pathological abnormalities, we found increased angiopoietin-2/angiopoietin-1 ratio and VEGF expression, which represent endothelial proliferation. Angiopoietin-1 is a Tie-2 agonist that protects against endothelial cell apoptosis to maintain homeostasis of the vasculature in PAH.[17]Angiopoietin-2 is an antagonist to Tie 2, blocking receptor activation by angiopoietin-1. Angiopoietin-2 participates in the formation of pathological neovascularization and vascular leakage together with VEGF by stimulating disordered endothelial cell proliferation,[6,18] and is up-regulated in plexiform lesions in idiopathic PAH patients.[18] VEGF is a specific mitogen of endothelial cells that contributes to the formation of plexiform lesions or endothelial cell clusters in PAH.[6] These results are in accordance with our present findings that angiopoietin-2 and VEGF were both expressed in plexiform lesions and VEGF was also expressed in concentric lesions by immunohistochemical analysis. Moreover, the protein expression of angiopoietin-2/angiopoietin-1 and VEGF increased in APE-CA by Western blotting analysis. These findings indicated that the activation of VEGF and increased angiopoietin-2/angiopoietin-1 ratio were associated with pulmonary arterial remodeling, which presented as plexiform and concentric lesions formed by disordered endothelial cell proliferation.
The classic ACE-Ang II-AT1 receptor axis of RAS is known to cause endothelial dysfunction.[19]ACE2-Ang (1-7)-Mas receptor axis opposed the detrimental effects of ACE-Ang II-AT1 receptor axis.[20] In a model of severe PAH induced by monocrotaline, neointimal occlusive lesions and impaired endothelium-dependent relaxation in pulmonary arteries were alleviated by ACE2.[21] However, it remains unclear the roles of pulmonary artery ACE2 and ACE axes in APE. In the present study, Western blotting analysis revealed increased ACE levels and reduced ACE2 levels in the pulmonary artery after CA due to APE. Immunohistochemistry revealed expression of ACE2 and Mas receptors in plexiform lesions after CA due to APE. The results indicated that ACE2/ACE axes imbalance participated in the pulmonary artery remodeling in APE.
The ACE inhibitor captopril infusion was associated with significantly lower mean right ventricular pressure and pulmonary vascular resistance, and higher serum Ang (1-7) levels, ACE2/ACE ratio, and Ang (1-7)/Ang II ratio after ROSC.[11] This study is an extension of previous research and we found that increasing ACE2/ACE axes ratio by captopril could alleviate pulmonary artery remodeling after ROSC caused by APE. The ACE axis has been shown to contribute to the activation of angiopoietin, VEGF, and pro-apoptotic factors. Otani et al[22]showed that Ang II induced the expression of angiopoietin-2, but not angiopoietin-1 or VEGF, in cultured bovine retinal endothelial cells. Madsen et al[23] found that AT1 receptor antagonism inhibited the transcription of VEGF, angiopoietin-1, angiopoietin-2, and Tie 2 in cortex and medulla in postnatal kidney injury and diabetic nephropathy.[24] In our investigation of the pulmonary artery remodeling in APE-CA, captopril alleviated histological endothelial cell proliferation and apoptosis, and reduced the angiopoietin-2/angiopoietin-1 ratio and VEGF levels as well as pro-apoptotic factor cleaved caspase-3 expression. The present findings might suggest a novel protective effect of increasing ACE2/ACE axes ratio on the inhibition of early pulmonary artery remodeling in APE. Our study had a certain limitation. The sample size is not enough to observe mortality.
CONCLUSIONS
The present findings show that the early pulmonary artery remodeling including endothelial cell proliferation and apoptosis occurs in massive APE pigs with CA. Increasing ACE2/ACE axes ratio reduces endothelial cell proliferation by inhibiting VEGF and angiopoietin-2/angiopoietin-1 ratio after ROSC. In addition, increasing ACE2/ACE axes ratio restrains endothelial cell apoptosis by reversing the imbalance in Bcl-2/Bax levels and decreasing cleaved caspase-3 expression.
Funding: This work was supported by grants from the National Natural Science Foundation of China (81773931 and 81374004); the Beijing Municipal Administration of Hospitals' Youth Program (QML20170105); the Natural Science Foundation of Beijing Municipality (7173253); and the Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support “Yangfan” Project (ZYLX201802).
Ethical approval: The experimental procedures were permitted by the Capital Medical University Institutional Animal Care Committee (permit number: 2010-D-013).
Conflicts of interest: No conflicts of interest declared.
Contributors: HLX: literature search, study design, animal care, data collection, data analysis, data interpretation, writing, and critical revision; LXZ: animal care, pathology methods, data collection, data analysis, and data interpretation; JY, NT, LA, and GXW: animal care, data collection, data analysis, and data interpretation; MRX and CSL: literature search, study design, data analysis, data interpretation, writing, and critical revision.
All the supplementary files in this paper are available at http://wjem.com.cn.
Reference
Saddle pulmonary embolism is not a sign of high-risk deterioration in non-high-risk patients: a propensity score-matched study
DOI:10.5847/wjem.j.1920-8642.2021.04.002 URL [Cited within: 1]
Characteristics and outcomes of cardiac arrest survivors with acute pulmonary embolism
DOI:10.1016/j.resuscitation.2020.06.029 URL [Cited within: 1]
Pathophysiology and treatment of haemodynamic instability in acute pulmonary embolism: the pivotal role of pulmonary vasoconstriction
PMID:11033105
[Cited within: 1]

Acute massive pulmonary embolism has a high mortality rate. Fatal haemodynamic deterioration is caused by an acute increase in pulmonary vascular resistance. Traditionally, the degree of mechanical obstruction of the pulmonary vasculature by the embolic thrombus is considered to be the major determinant of this increase in right ventricular afterload. However, there is evidence to suggest that another factor plays an important role, since there is a marked discrepancy between the haemodynamic manifestations of acute pulmonary embolism and the degree of mechanical obstruction. Historic studies indicate that this discrepancy is largely explained by pulmonary vasoconstriction caused by vasoactive mediators, released mainly by activated platelets. Thromboxane-A(2) and serotonin are probably the two most important pulmonary vasoconstrictors in this context. Antagonising their effects dramatically increases tolerance to experimental pulmonary embolism in animals. In humans, this concept should eventually find its way into clinical practice. In the future, acute massive pulmonary embolism could be treated with antagonists to pulmonary vasoconstrictors, or with direct pulmonary vasodilators.
Pulmonary thromboendarterectomy: a clinicopathologic study of 200 consecutive pulmonary thromboendarterectomy cases in one institution
PMID:17350667
[Cited within: 1]
Approximately 2000 patients underwent pulmonary thromboendarterectomy (PTE) to date at University of California, San Diego. We retrospectively reviewed the clinicopathologic manifestations of 200 consecutive PTE cases from June 2004 to February 2006 with an emphasis on the histopathologic spectrum of chronic thromboembolic disease. Pathology reports and all histologic sections of study cases were examined. Pertinent clinical data were obtained from operative reports and medical records. In the study group, there were 2 cases (1 man, 1 woman) of pulmonary artery sarcomas and 1 case of metastatic tumor emboli from a testicular germ cell tumor. Two patients (both women) showed histologic evidence of arteritis without clinically apparent systemic vasculitis. The remaining 195 PTE patients with chronic thromboembolic disease consisted of 97 women and 98 men with a mean age of 52 (range, 17-83) and 51 (range, 16-82), respectively. Bilateral PTE was performed in 191, and unilateral PTE was performed in 4 (right and left, 2 each) patients. History of deep vein thrombosis was noted in 38.5%, and coagulation abnormalities were documented in 16.4% of these 195 cases. Grossly, the volumes of PTE specimens were greater in men than in women and on the right side than on the left in both men and women. Microscopically, the thrombi were recent fibrinous clot in 0.8%, mixed fibrinous and organizing in 45%, and old organized in 54.2% of specimens. Inflammation within the thrombi was usually mild but moderate and severe inflammation was found in 13.4% and 1.3% of specimens, respectively. Exuberant epithelioid granulomas were seen within the thrombi in one patient who had a history of sarcoidosis. Collections of foamy histiocytes and/or cholesterol clefts were found in 45%, and calcification was present in 11.5% of specimens. One case revealed diffuse myofibroblastic proliferation in a highly inflammatory background containing numerous plasma cells, reminiscent of inflammatory myofibroblastic tumor. In summary, pathology of PTE specimens in our study group encompassed remodeling of thrombi at various stages with variable degrees of inflammation and cellularity, granulomas associated with sarcoidosis, a rare case showing features of inflammatory myofibroblastic tumor, primary or metastatic malignancy, and isolated pulmonary arteritis.
Endothelial dysfunction in pulmonary hypertension
PMID:14734504 [Cited within: 4]
Cellular and molecular basis of pulmonary arterial hypertension
Expression of pulmonary vascular angiotensin-converting enzyme in primary and secondary plexiform pulmonary hypertension
PMID:11054722
[Cited within: 1]
The hypothesis for this study was that increased local expression of vascular angiotensin-converting enzyme (ACE) may contribute to the arterial remodelling which accompanies pulmonary hypertension, since angiotensin II (ANG II) is an important mediator of pulmonary vascular cell growth. The expression of ACE was studied by immunohistochemistry in paraffin-embedded lung sections from adults undergoing heart-lung transplantation for severe primary (n=6) and secondary (n=7) pulmonary arterial hypertension (PH), compared with age-matched controls (n=11). An antigen retrieval technique was used prior to incubating sections with the anti-ACE monoclonal antibody, CG2, or the endothelial marker, monoclonal anti-CD31. In control lungs, the highest level of ACE immunostaining was seen in the alveolar capillary endothelium, with less intense staining in small intra-acinar pulmonary arteries and relatively little staining in larger preacinar arteries. ACE immunostaining was virtually absent in lymphatics and veins. In both primary and secondary PH, there was an increase in ACE immunostaining in the endothelium of intra-acinar peripheral pulmonary arteries compared with control lungs, extending to the level of alveolar ducts, as confirmed by semi-quantitative analysis. The increase in endothelial ACE expression in the intra-acinar arteries of patients with primary and secondary PH is consistent with the hypothesis that locally increased production of ANG II may contribute to the process of pulmonary vascular remodelling.Copyright 2000 John Wiley & Sons, Ltd.
Angiotensin-converting enzyme 2 inhibits apoptosis of pulmonary endothelial cells during acute lung injury through suppressing SMAD2 phosphorylation
DOI:10.1159/000374025 URL [Cited within: 1]
Perspectives for angiotensin profiling with liquid chromatography/mass spectrometry to evaluate ACE/ACE2 balance in endothelial dysfunction and vascular pathologies
DOI:10.1016/j.pharep.2015.03.017
PMID:26321281
[Cited within: 1]
Vascular injury, characterized by endothelial dysfunction, inflammation, structural remodeling, thrombosis and calcification leads to cardiovascular diseases. Angiotensin (Ang) II (1-8) - synthesized mainly by angiotensin converting enzyme (ACE) is the best characterized mediator of the renin-angiotensin system (RAS). This peptide initially identified by its vasoactive properties was found to play a major role in vascular response to insult. However, recent discovery of angiotensin converting enzyme 2 (ACE2) that produces vasoprotective Ang-(1-7) peptide highlighted complexity of the system and suggested that balance between ACE/Ang II and ACE2/Ang-(1-7) is fundamental in maintaining vascular homeostasis and its disorders are associated with cardiovascular pathology. There is therefore a need to develop methods for comprehensive analysis of biologically active Ang peptides and their metabolites of ACE/Ang II and ACE2/Ang-(1-7) axes. Liquid chromatography/mass spectrometry (LC/MS) is an analytical technique that offers potential for specific, simultaneous analysis of Ang peptides. With sensitivity added by application of preconcentration nanochromatography reaching picomolar concentrations, practically all Ang peptides identified so far could be quantified in biological samples. Ang profiling is important not only for understanding their physiological or pathological role but could also serve as an early diagnostic biomarker of endothelial dysfunction and cardiovascular pathology. It could also be used for monitoring the efficacy of the RAS-targeted therapies. Although, the methodology requires further improvements to adopt it for routine application, Ang peptide profiling with targeted LC/MS analysis might assess functional balance between ACE/Ang II and ACE2/Ang-(1-7) axes, facilitate our understanding of the cardiovascular pathology and enhance biomarker portfolio in cardiovascular diseases. Copyright © 2015 Institute of Pharmacology, Polish Academy of Sciences. Published by Elsevier Urban & Partner Sp. z o.o. All rights reserved.
Captopril improves postresuscitation hemodynamics protective against pulmonary embolism by activating the ACE2/Ang-(1-7)/Mas axis
DOI:10.1007/s00210-016-1278-7 URL [Cited within: 2]
Use of the impedance threshold device improves survival rate and neurological outcome in a swine model of asphyxial cardiac arrest
DOI:10.1097/CCM.0b013e318232d8de
PMID:21983368
[Cited within: 2]
To assess whether intermittent impedance of inspiratory gas exchange improves hemodynamic parameters, 48-hr survival, and neurologic outcome in a swine model of asphyxial cardiac arrest treated with active compression-decompression cardiopulmonary resuscitation.Prospective, randomized, double-blind study.Laboratory investigation.Thirty healthy Landrace/Large-White piglets of both sexes, aged 10 to 15 wks, whose average weight was 19 ± 2 kg.At approximately 7 mins following endotracheal tube clamping, ventricular fibrillation was induced and remained untreated for another 8 mins. Before initiation of cardiopulmonary resuscitation, animals were randomly assigned to either receive active compression-decompression cardiopulmonary resuscitation plus a sham impedance threshold device (control group, n = 15), or active compression-decompression cardiopulmonary resuscitation plus an active impedance threshold device (experimental group, n = 15). Electrical defibrillation was attempted every 2 mins until return of spontaneous circulation or asystole.Return of spontaneous circulation was observed in six (40%) animals treated with the sham valve and 14 (93.3%) animals treated with the active valve (p =.005, odds ratio 21.0, 95% confidence interval 2.16-204.6). Neuron-specific enolase and S-100 levels increased in the ensuing 4 hrs post resuscitation in both groups, but they were significantly elevated in animals treated with the sham valve (p <.01). At 48 hrs, neurologic alertness score was significantly better in animals treated with the active valve (79.1 ± 18.7 vs. 50 ± 10, p <.05) and was strongly negatively correlated with 1- and 4-hr postresuscitation neuron-specific enolase (r = -.86, p <.001 and r = -.87, p <.001, respectively) and S-100 (r = -.77, p <.001 and r = -0.8, p =.001) values.In this model of asphyxial cardiac arrest, intermittent airway occlusion with the impedance threshold device during the decompression phase of active compression-decompression cardiopulmonary resuscitation significantly improved hemodynamic parameters, 24- and 48-hr survival, and neurologic outcome evaluated both with clinical and biochemical parameters (neuron-specific enolase, S-100).
The effects of nifedipine on ventricular fibrillation mean frequency in a porcine model of prolonged cardiopulmonary resuscitation
DOI:10.1213/01.ane.0000068801.28430.ed
PMID:12818971
[Cited within: 1]
We assessed the effects of a calcium channel blocker versus saline placebo on ventricular fibrillation mean frequency and hemodynamic variables during prolonged cardiopulmonary resuscitation (CPR). Before cardiac arrest, 10 animals were randomly assigned to receive either nifedipine (0.64 mg/kg; n = 5) or saline placebo (n = 5) over 10 min. Immediately after drug administration, ventricular fibrillation was induced. After 4 min of cardiac arrest and 18 min of basic life support CPR, defibrillation was attempted. Ninety seconds after the induction of cardiac arrest, ventricular fibrillation mean frequency was significantly (P < 0.01) increased in nifedipine versus placebo pigs (mean +/- SD: 12.4 +/- 2.1 Hz versus 8 +/- 0.7 Hz). From 2 to 18.5 min after the induction of cardiac arrest, no differences in ventricular fibrillation mean frequency were detected between groups. Before defibrillation, ventricular fibrillation mean frequency was significantly (P < 0.05) increased in nifedipine versus placebo animals (9.7 +/- 1.2 Hz versus 7.1 +/- 1.3 Hz). Coronary perfusion pressure was significantly lower in the nifedipine than in the placebo group from the induction of ventricular fibrillation to 11.5 min of cardiac arrest; no animal had a return of spontaneous circulation after defibrillation. In conclusion, nifedipine, but not saline placebo, prevented a rapid decrease of ventricular fibrillation mean frequency after the induction of cardiac arrest and maintained ventricular fibrillation mean frequency at approximately 10 Hz during prolonged CPR; this was nevertheless associated with no defibrillation success.This study evaluates the effects of a calcium channel blocker on ventricular fibrillation mean frequency, hemodynamic variables, and resuscitability during prolonged cardiopulmonary resuscitation (CPR) in pigs. Nifedipine, but not saline placebo, prevented a rapid decrease of ventricular fibrillation mean frequency after the induction of cardiac arrest and maintained ventricular fibrillation mean frequency at approximately 10 Hz during prolonged CPR but did not improve resuscitability.
Captopril suppresses inflammation in endotoxin-induced uveitis in rats
DOI:10.1016/j.exer.2006.03.005 URL [Cited within: 1]
ROS scavenger decreases basal perfusion pressure, vasoconstriction and NO synthase activity in pulmonary circulation during pulmonary microembolism
PMID:25804094
[Cited within: 1]
Two mechanisms contribute in the development of pulmonary hypertension in pulmonary embolism (PE) - obstruction of pulmonary blood vessels and vasoconstriction. We hypothesize that hypoxia, increased shear stress and/or activation of gathered leukocytes in the PE may cause a release of reactive oxygen species (ROS). Therefore our aim was to determine the influence of the ROS scavenger Tempol on pulmonary hypertension and to describe NO synthase activity and production of NO oxidative products (NOx) after PE. In general anesthesia sephadex microspheres suspended in PSS were applied in right jugular vein as the pulmonary microembolism. Than we measured in isolated salt solution-perfused lungs the changes in perfusion pressure, activity of NO synthase and NOx plasma concentration in 7 groups of rats: C: control group (n=5), CN: C + sodium nitroprusside (SN) (n=5), EN: PE + SN (n=5), ETN: Tempol + PE + SN (n=5), CL: C + L-NAME (n=5), EL: PE + L-NAME (n=5), ETL: Tempol + PE + L-NAME (n=5). Tempol was applied intraperitoneally before PE. Animals that received Tempol (groups TN, TL) had significantly lower basal perfusion pressure than those which did not receive Tempol (EN, EL). Overall we measured a higher decrease of perfusion pressure than in the control group (C) after application of SN. Administration of L-NAME after PE (EL) increased the pressure more than in the control group (NL). NOx concentration was higher after PE. We found that preventive administration of Tempol decreases the increase in perfusion pressure after PE. PE increased NO release and concentration of NOx.
Pathology of pulmonary hypertension
DOI:10.1016/j.ccm.2013.08.009 URL [Cited within: 1]
The angiopietin-1-Tie2 pathway prevents rather than promotes pulmonary arterial hypertension in transgenic mice
DOI:10.1084/jem.20090389 URL [Cited within: 1]
Circulating angiopoietins in idiopathic pulmonary arterial hypertension
DOI:10.1093/eurheartj/ehq226 URL [Cited within: 2]
Endothelial dysfunction: the role and impact of the renin-angiotensin system
Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension
DOI:10.1164/rccm.200811-1678OC URL [Cited within: 1]
ACE2 activation confers endothelial protection and attenuates neointimal lesions in prevention of severe pulmonary arterial hypertension in rats
DOI:10.1007/s00408-013-9470-8 URL [Cited within: 1]
Angiotensin II induces expression of the Tie2 receptor ligand, angiopoietin-2, in bovine retinal endothelial cells
DOI:10.2337/diabetes.50.4.867
PMID:11289054
[Cited within: 1]
Recent studies have shown that angiopoietins (Angs) and their receptor, Tie2, play a role in vascular integrity and neovascularization. The renin-angiotensin system has been hypothesized to contribute to the development of diabetic retinopathy. In this study, we investigated the effect of angiotensin II (AII) on Ang1 and Ang2 expression in cultured bovine retinal endothelial cells (BRECs). AII stimulated Ang2 but not Ang1 mRNA expression in a dose- and time-dependent manner. This response was inhibited completely by angiotensin type 1 receptor (AT1) antagonist. AII increased the transcription of Ang2 mRNA, but did not change the half-life. Protein kinase C (PKC) inhibitor completely inhibited AII-induced Ang2 expression, and the mitogen-activated protein kinase (MAPK) inhibitor also inhibited it by 69.4+/-15.6%. In addition, we confirmed the upregulation of Ang2 in an AII-induced in vivo rat corneal neovascularization model. These data suggest that AII stimulates Ang2 expression through AT1 receptor-mediated PKC and MAPK pathways in BREC, and AII may play a novel role in retinal neovascularization.
Angiotensin II promotes development of the renal microcirculation through AT1 receptorsv
DOI:10.1681/ASN.2009010045 URL [Cited within: 1]
Temporal renal expression of angiogenic growth factors and their receptors in experimental diabetes: role of therenin-angiotensin system
PMID:15643138
[Cited within: 1]
It has been postulated that vascular endothelial growth factor (VEGF) plays a role in the progression of renal injury. However, the role of other angiogenic factors and their receptors, such as the angiopoietins and Tie2, and in particular their relation to renoprotective therapies, such as agents that interrupt the renin-angiotensin system, have not been studied in the context of diabetes-related renal injury.Renal expression of VEGF, angiopoietin-1 (Ang-1), angiopoietin-2 (Ang-2) and their receptors, VEGF-R2 and Tie-2, were assessed using reverse transcription-polymerase chain reaction, immunohistochemistry and Western blotting, in control and streptozotocin diabetic rats, untreated or receiving the AT1 receptor antagonist, valsartan, or the AT2 receptor antagonist, PD123319.Diabetes was associated with increased gene and protein expression of VEGF, VEGF-R2, Ang-1, Ang-2 and Tie-2. AT1 receptor antagonism attenuated gene expression of these cytokines and receptors, yet PD123319, which had no effect on blood pressure, reduced VEGF-R2 and Ang-1 gene expression and decreased VEGF, Ang-1 and Ang-2 protein levels.In experimental diabetes, there is significant upregulation within the kidney of various angiogenic cytokines and their receptors. Furthermore, the effects of angiotensin II receptor blockade on these parameters is consistent with the VEGF-VEGF-R2 and angiopoietin-Tie-2 axes being modulated in the kidney by haemodynamic factors in the diabetic context.

{kind=link}
{kind=link}
{kind=link}
{kind=link}