


Lipid storage myopathy (LSM) is a manifestation of lipid dysmetabolism, presenting with lipid accumulation in muscles. The mechanism includes defects in intracellular triglyceride catabolism, transport of long-chain fatty acids and carnitine, or fatty acid β-oxidation.[1] Among LSMs, the most common type is multiple acyl-coenzyme A dehydrogenase deficiency (MADD), also called glutaric aciduria type II (GA II), with an estimated prevalence of 1 in 20,000 to 1 in 15,000 births in the United States.[2] A cohort study of 90 Chinese patients showed that the most frequent mutations of MADD were c.250G>A, c.770A>G, and c.1227A>C in the electron transfer flavoprotein dehydrogenase (ETFDH) gene.[3]Common symptoms include symmetrical weakness of the proximal upper and/or lower limbs, steatohepatitis, ketoacidosis, and hypoglycemia.[4,5] However, cases with both respiratory and cardiac muscles affected have rarely been reported. Here, we report a case of a girl who presented with increased serum enzyme levels, labored breathing, cardiogenic shock, and eventual cardiac arrest, which is a rare and lethal complication of GA II. Informed consent for publication was obtained from the patient’s family.
A 15-year-old girl was transferred to the intensive care unit (ICU) following initial cardiopulmonary resuscitation (CPR). She had a medical history of chronic pancreatitis for three years, with three to four attacks per year. She was admitted to the gastroenterology ward due to acute recurrent pancreatitis, hepatic injury, and mild weakness of the proximal lower limbs. Then, she rapidly developed muscle weakness with shallow breathing, elevated serum muscle enzyme levels, rhabdomyolysis, and cardiogenic shock, resulting in cardiac arrest. She underwent tracheal intubation and CPR and achieved successful restoration of spontaneous circulation (ROSC) within 10 min. She was hemodynamically unstable, receiving continued norepinephrine (3 µg/[kg·min]) and dobutamine (5 µg/[kg·min]) infusion following ROSC. She was subsequently admitted to the ICU for further treatment.
In the ICU, vasoactive drugs were maintained for over 24 h and then gradually reduced until complete withdrawal. Since the electrical activity of the diaphragm (Edi) was low, mechanical ventilation was maintained for two weeks prior to weaning. She developed cholestatic jaundice and worsening hepatocellular dysfunction. Ultrasound and liver biopsy met the diagnosis of steatohepatitis. Echocardiography showed hypertrophy in the left ventricle and interventricular septum with decreased echo (Figure 1 A). Magnetic resonance imaging showed atrophy and abnormal signals in the neck and lower limb muscles (Figures 1 C and D). Electromyography indicated myogenous damage.
Figure 1.
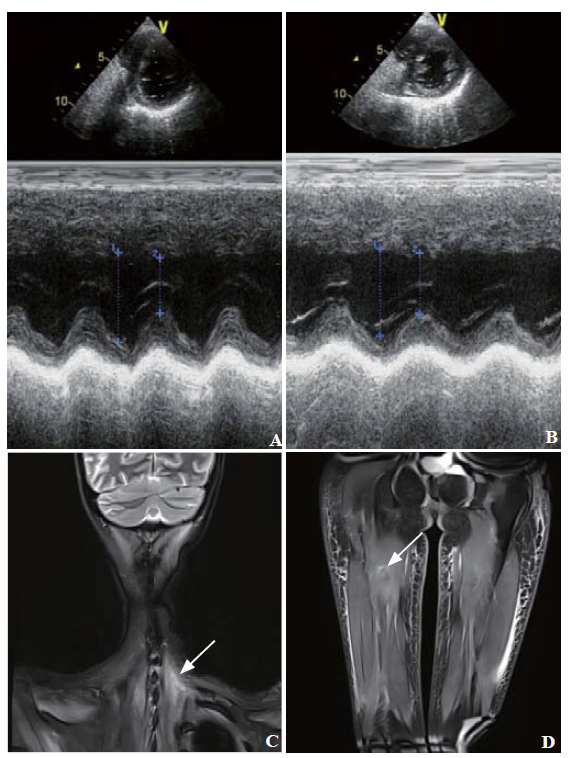
Figure 1.
Echocardiography and magnetic resonance imaging.Echocardiography findings: images before treatment (A) and one year later (B); magnetic resonance imaging of the neck and lower limbs showed soft tissue edema, atrophy and abnormal signals of muscles: T2 fat suppression imaging of the neck (C), proton fat suppression imaging of the lower limbs (D). Arrow: muscle injury.
For differential diagnosis, mass spectroscopy of serum samples was performed, revealing a high level of predominantly middle- and long-chain acyl carnitines (Table 1, supplementary Table 1, and supplementary Figure 1). However, urinary organic acids were normal (supplementary Figure 2). Thus, muscle biopsy and genetic testing were performed. Under a light microscope, muscle fibers showed focally mild-to-moderate atrophy, accompanied by myolysis and necrosis with phagocytosis. The muscle also showed mild proliferation of interstitial fibers. Numerous vacuoles of very different sizes were observed in muscle fibers (Figure 2A). Nicotinamide adenine dinucleotide (NADH) staining and adenosine triphosphatase (ATPase) staining showed that these vacuoles mainly existed in type I fibers (Figures 2 B and C); Oil Red O staining demonstrated that the intracellular vacuoles were lipid droplets (Figure 2D). These findings were consistent with those of myogenous damage, indicating lipid storage myopathy with myofiber necrosis. Under an electron microscope, a medium-large number of lipid droplets were observed in muscle fibers (Figure 2 E). Whole exome sequencing of serum samples showed two pathogenic mutations in the ETFDH gene, with two heterozygote missense mutations, c.250G>A (p. Ala84Thr) in exon 3 and c.524G>A (p. Arg175His) in exon 5, and one suspected pathogenic mutation in the protease serine 1 (PRSS1) gene c.346C>T (p. Arg116Cys) in exon 3 (supplementary Table 1).
Table 1. Results of tandem mass spectroscopy in serum sample
| Parameters | Results, μmol/L | Reference index, μmol/L |
|---|---|---|
| Octanoyl carnitines (C8) | 0.41 | 0.01-0.30 |
| Decanoyl carnitines (C10) | 0.57 | 0.01-0.40 |
| Palmitoyl carnitines (C16:1) | 0.41 | 0.02-0.30 |
| C3DC/C4 | 0.08 | 0.10-1.50 |
| C5-OH/C8 | 0.26 | 0.40-25.00 |
Figure 2.
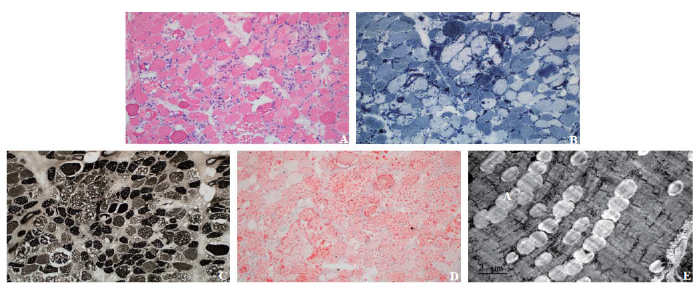
Figure 2.
Histopathological changes in skeletal muscles. A: hematoxylin and eosin staining (×200); B: nicotinamide adenine dinucleotide (NADH) stain (×200); C: adenosine triphosphatase (ATPase) stain (×200); D: Oil Red O stain (×200); E: electron microscope image (×200).
Based on these findings, a diagnosis of GA II was made. She was given a high-carbohydrate, low-fat, and low-protein diet with riboflavin (150 mg/day orally), coenzyme Q10 (CoQ10) (300 mg/day orally), and L-carnitine (1,000 mg/ day intravenously) supplementation. Muscle weakness was improved during withdrawal of positive pressure ventilation. Meanwhile, pancreatitis and hepatic injury were cured. She was eventually discharged without any neurological sequelae of cardiac arrest on day 27 of hospitalization. By the one-year follow-up, she presented with normal muscle strength, cardiac enzymes and echocardiography (Figure 1 B) and no attack of pancreatitis.
In this case, the patient presented with progressive muscle weakness, especially involving the respiratory and cardiac muscles, accompanied by severe steatohepatitis and recurrent pancreatitis. In muscle biopsy, lipid accumulation, muscle atrophy, and fiber necrosis were identified. Tandem mass spectroscopy of serum samples revealed elevated levels of middle- and long-chain acyl carnitines, accounting for the diagnosis of LSM. Due to normal levels of organic acids in urine, gene testing was performed, showing two pathogenic mutations in the ETFDH gene, further confirming the MADD diagnosis. The diagnoses of steroid myopathy and mitochondrial myopathy were excluded, considering the normal result of mitochondrial genetic testing and the absence of steroid usage.
During the first 2 weeks of her ICU stay, she was supported by mechanical ventilation because of a low Edi value, suggesting damage to the muscles. Combining the results of echocardiography, we assumed the possibility of cardiomyopathy, which has only been reported in few patients.[6,7] We should highlight that patients with rapidly progressive respiratory muscle weakness and cardiomyopathy have a relatively higher risk of cardiac arrest, requiring prompt treatment.
The combination of pancreatitis attacks and a suspected pathogenic mutation in the PRSS1 gene, one of the most common pathogenic variations of hereditary pancreatitis,[8⇓-10] should lead to considerations of hereditary pancreatitis. To date, there have been no reports of hereditary pancreatitis associated with LSM. However, pancreatitis may also be a rare complication of LSM, with unknown mechanisms. The first case of acute pancreatitis in MADD was reported in 1997.[11] To date, only one case of recurrent pancreatitis in MADD was reported in 2004, without detecting pancreatic gene mutations.[12] LSM is characterized by lipid accumulation in muscles, including smooth muscles; thus, it is speculated that the Oddi sphincter was affected in this patient. In this case, no recurrence of pancreatitis during the one-year follow-up confirmed that Oddi sphincter dysfunction might be a complication of GA II despite a genetic predisposition. The function of the Oddi sphincter could be tested through quantitative hepatobiliary scintigraphy, secretin-stimulated magnetic resonance cholangiopancreatography, or sphincter of Oddi manometry.[13⇓-15] However, the limitation without further detection in our case is due to the critical condition of the patient.
In our case, cardiac arrest resulted from myopathy in cardiac and respiratory muscle. Early biopsy and genetic analysis can contribute to the accurate diagnosis of GA II, especially in patients with ambiguous mass spectroscopy results. Recurrent pancreatitis might be a rare complication owing to lipid accumulation in the Oddi sphincter.
Funding: This study was supported by the Youth Projects of National Natural Science Foundation of China (81601662) and the Medical Innovation Program of Fujian Province of China (2020CXA003).
Ethical approval: Informed consent for publication was obtained from the patient’s family.
Conflicts of interest: None.
Contributors: HPX and WJZ contributed equally to this work as co-first authors. HPX and WJZ proposed the study and wrote the paper. All authors contributed to the design and interpretation of the study and to further drafts.
All the supplementary files in this paper are available at http://wjem.com.cn.
Reference
Lipid storage myopathy
DOI:10.1007/s11910-010-0154-y URL [Cited within: 1]
Riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency in 13 cases, and a literature review in Chinese mainland patients
DOI:10.1038/jhg.2014.10 URL [Cited within: 1]
Clinical features and ETFDH mutation spectrum in a cohort of 90 Chinese patients with late-onset multiple acyl-CoA dehydrogenase deficiency
DOI:10.1007/s10545-013-9671-6 URL [Cited within: 1]
Multiple acyl-CoA-dehydrogenase deficiency (MADD)—a novel mutation of electron-transferring-flavoprotein dehydrogenase ETFDH
DOI:10.1016/j.jns.2011.05.001
PMID:21616504
[Cited within: 1]

This is the case of a 41 year old man, suffering general weakness and elevated liver enzymes, sensitive to a treatment with riboflavin and coenzyme Q(10). Tandem mass spectroscopy and molecular analysis reveal a multiple acyl-CoA-dehydrogenase deficiency (MADD) with two novel heterozygote missense mutations of the EFTDH gene.Copyright © 2011 Elsevier B.V. All rights reserved.
Lipid storage myopathies: current treatments and future directions
DOI:S0163-7827(18)30017-1
PMID:30099045
[Cited within: 1]
Lipid storage myopathies (LSMs) are a heterogeneous group of genetic disorders that present with abnormal lipid storage in multiple body organs, typically muscle. Patients can clinically present with cardiomyopathy, skeletal muscle weakness, myalgia, and extreme fatigue. An early diagnosis is crucial, as some LSMs can be managed by simple nutraceutical supplementation. For example, high dosage l-carnitine is an effective intervention for patients with Primary Carnitine Deficiency (PCD). This review discusses the clinical features and management practices of PCD as well as Neutral Lipid Storage Disease (NLSD) and Multiple Acyl-CoA Dehydrogenase Deficiency (MADD). We provide a detailed summary of current clinical management strategies, highlighting issues of high-risk contraindicated treatments with case study examples not previously reviewed. Additionally, we outline current preclinical studies providing disease mechanistic insight. Lastly, we propose that a number of other conditions involving lipid metabolic dysfunction that are not classified as LSMs may share common features. These include Neurofibromatosis Type 1 (NF1) and autoimmune myopathies, including Polymyositis (PM), Dermatomyositis (DM), and Inclusion Body Myositis (IBM).Crown Copyright © 2018. Published by Elsevier Ltd. All rights reserved.
Cardiomyopathy in multiple acyl-CoA dehydrogenase deficiency: a clinico-pathological correlation and review of literature
DOI:10.1007/s00246-007-9119-6
PMID:17912479
[Cited within: 1]
Multiple acyl-CoA dehydrogenase deficiency (MADD) is a rare autosomal recessive defect of the electron transfer flavoprotein or ubiquinone oxidoreductase, resulting in abnormal fatty acid, amino acid, and choline metabolism, leading to metabolic acidosis, hypoglycemia, "sweaty-feet" odor, and early neonatal deaths. This report presents a child diagnosed with this disease at birth by newborn screening using the mass spectrometer, who died suddenly at the age of 6 months. The echocardiogram revealed pericardial effusion, thickened ventricular musculature, and insufficiency of both the atrio-ventricular valves. The autopsy showed immense cardiomegaly, fatty infiltration, and hypertrophy of the ventricles. This is the first detailed case report of clinico-pathological correlation of MADD in an infant and brings into light a rare form of cardiomyopathy as a differential diagnosis in critically ill patients.
Late-onset multiple acyl-CoA dehydrogenase deficiency with cardiac syncope: a case report
The role of genetics in pancreatitis
DOI:S1052-5157(18)30728-1
PMID:30241646
[Cited within: 1]
Individuals with acute recurrent and chronic pancreatitis may have an inherited predisposition to the development of the disease. Pancreatitis in the setting of a significant family history of the disease can be classified as hereditary or familial pancreatitis. In this article, the authors closely examine the specific genes implicated in pancreatitis, investigate the role of genetic testing for diagnosis, and describe the impact of genetic testing results on clinical management.Copyright © 2018 Elsevier Inc. All rights reserved.
Hereditary pancreatitis: current perspectives
DOI:10.2147/CEG.S84358
PMID:27555793
[Cited within: 1]
Hereditary pancreatitis (HP) is a rare cause of acute, recurrent acute, and chronic pancreatitis. It may present similarly to other causes of acute and chronic pancreatitis, and often there has been a protracted evaluation prior to the diagnosis of HP. Since it was first described in 1952, multiple genetic defects that affect the action of digestive enzymes in the pancreas have been implicated. The most common mutations involve the PRSS1, CFTR, SPINK1, and CTRC genes. New mutations in these genes and previously unrecognized mutations in other genes are being discovered due to the increasing use of next-generation genomic sequencing. While the inheritance pathways of these genetic mutations may be variable and complex, sometimes involving coinheritance of other mutations, the clinical presentation of patients tends to be similar. Interactions with environmental triggers often play a role. Patients tend to present at an early age (prior to the second decade of life) and have a significantly increased risk for the development of pancreatic adenocarcinoma. Patients with HP may develop sequelae of chronic pancreatitis such as strictures and fluid collections as well as exocrine and endocrine insufficiency. Management of patients with HP involves avoidance of environmental triggers, surveillance for pancreatic adenocarcinoma, medical therapy for endocrine and exocrine insufficiency, pain management, and endoscopic or surgical treatment for complications. Care for affected patients should be individualized, with an emphasis on early diagnosis and multidisciplinary involvement to develop a comprehensive treatment strategy.
Increased activation of hereditary pancreatitis-associated human cationic trypsinogen mutants in presence of chymotrypsin C
Acute pancreatitis in a patient with glutaric acidemia type II
Riboflavin-responsive glutaric aciduria type II with recurrent pancreatitis
DOI:10.1016/j.pediatrneurol.2004.02.015 URL [Cited within: 1]
Assessment of the reproducibility of quantitative hepatobiliary scintigraphy (QHBS) in patients with sphincter of Oddi dysfunction (SOD)—inappropriate method or intermittent disease?
The effects of morphine-neostigmine and secretin provocation on pancreaticobiliary morphology in healthy subjects: a randomized, double-blind crossover study using serial MRCP
DOI:10.1007/s00268-011-1162-z
PMID:21656308
[Cited within: 1]
Secretin-stimulated magnetic resonance cholangiopancreatography (MRCP) is used for the diagnosis of sphincter of Oddi dysfunction (SOD), but it does not correlate well with sphincter of Oddi manometry. Serial MRCP following morphine-neostigmine provocation may be of value in the assessment of SOD, but the effects of these pharmacological agents on pancreaticobiliary morphology in healthy subjects have not been studied. The aim of the present study was to use serial MRCP to characterize the effects of morphine-neostigmine and secretin provocation on serum pancreatic enzyme responses and pancreaticobiliary ductal morphology in healthy subjects.Following a baseline scan and serum lipase and amylase assays, 10 healthy subjects were randomized in a double-blind manner to receive morphine (10 mg intramuscularly [IM]), neostigmine (1 mg IM) and saline (intravenously [IV]); OR saline (IM), saline (IM) and secretin (1 U/kg IV). A MRCP study was performed at 5, 30, 60, 90, 120, 150, and 180 min thereafter, with blood samples taken every 60 min for 4 h. Pancreatic duct (PD) diameter, visible PD length, common bile duct (CBD) diameter, and gallbladder volume were recorded. Crossover studies were performed 10 days later.Serum pancreatic enzyme concentrations were significantly greater (amylase, P = 0.003; lipase, P = 0.04) after morphine-neostigmine than after secretin. Following morphine-neostigmine and secretin provocation, the mean (SEM) percentage increase in PD diameter was 28.7 (7.2) versus 12.9 (3.3); P < 0.0001, and visible PD length was 49.4 (11.5) versus 28.1 (8.2); P < 0.0001, respectively.The effects of morphine-neostigmine were more pronounced than those of secretin in healthy subjects. The diagnostic utility of morphine-neostigmine stimulated serial MRCP for SOD merits further evaluation.
A new manometry device for evaluating the sphincter of Oddi using a fiber-optic pressure sensor
DOI:10.1080/13645706.2017.1412701 URL [Cited within: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}